ANTI-ASTHMATIC AGENTS
Bronchial asthma is a hypersensitivity reaction of the lower respiratory system producing bronchoconstriction, muscosal edema, and excess mucus. A common end point of asthma is bronchial narrowing which gives rise to the difficulty in breathing and wheezing that are characteristic of airway obstruction. Asthma is noncotagious but affects nearly 9 million people of all ages in India. Children may suffer asthma episodes at very early ages and continue having them throughout their lives, or they may outgrow their asthma when they reach adolescence. Others develop asthma late in life. The asthmatic episodes may be continuous or sporadic with slight to severe impairment of respiratory function. An episode may affect normal life functioning only slightly, or may produce a high degree of disability. The individual may recover spontaneously or through proper drug therapy. Each year as many as 3000 deaths occur in the United States as result of asthma. Although asthma is normally not fatal, the chronic and unpredictable nature of the episodes are quite debilitating.
There are several methods of classifying asthma. One of the earliest and most popular methods is to categorize clinical asthma into extrinsic asthma, sometimes called allergic or atopic asthma, and intrinsic asthma, also termed idiopathic or nonatopic asthma. In extrinsic asthma causal allergens are demonstratable; in intrinsic asthma they are not. An extrinsic asthmatic usually has a family history of allergies. The allergen may include food, drugs, wood, feathers, pollens, mold spores, cosmetics, animal danders, hairs, house dust, and a variety of other foreign materials. These allegens, harmless to a normal individual, can initiate an attack in an asthmatic. If the allergen is placed in the skin, one normally sees a positive reaction to the allergen with inflammation, swelling, and redness occurring. The onset of extrinsic asthma is normally during childhood or adolesence, and elevated levels of specific antibodies, eg, immunoglobulin E (IgE), are usually observed in extrinsic asthmatic.
Intrisic asthma often begins in adulthood, and it appears allergens do not play a role in the etiology. Attacks of intrinsic asthma may be related to infection, exercise and/or psychological stress. Some individuals may have mixed asthma, a combination of extrinsic and intrinsic asthma: thus a clear cut distinction cannot always be made. Also, a child may have extrinsic asthma and after becoming and adult develop intrinsic asthma. It is difficult for some individuals to decide the relative contribution of psychogenic, infective, allergic, or exercise-induced factors in an asthmatic patient and the relative importance of each of these factors in the final clinical condition. In some patients their relative importance varies with time. Because extrinsic and intrinsic asthma episodes are so similar, it is difficult to distinguish between them during an acute attack.
The fundamental causes of asthma are still unknown. The management of patients is usually thought of in terms of removing the trigger mechanism if possible (3). In extrinsic asthma the attack can be prevented by removing the source of the allergen. However, in some instances, eg with mold spores and pollen, it is very difficult to remove the source. Intrinsic asthma shows a greater tendency to become chronic with development of coughs and sputum between acute episodes (2).
At present there is no known cure for the fundamental abnormality in asthma. Treatment of asthma involves the use of drugs for the prevention or relief of symptoms of asthma. If it is not possible to avoid the allergen, treatment of the patient with small graded doses of a known antigen at set intervals, called hyposensitization or desensitization, is successful in some instances. Although it is impossible to predict which patients will be helped by hyposensitization, this treatment is usually worth trying in patients who have a chronic disability due to asthma and when other methods of treatment have failed.
The principal aim in the management if asthma is to maintain the patient's normal pulmonary status for as much of the time as possible. It is exceedingly important to find drugs that will relieve or prevent the acute or chronic patterns of asthma, and that will also minimize the side effects that may occur. The method and ease of administration and the duration of action are important factors in developing drugs to treat asthma. This article deals with the following types of known or proposed anti-asthmatic agents: adrenergics stimulant; anticholinergic; inhibitors of the release of allergic mediators: xanthine derivatives; prostaglandins; and miscellaneous drugs.
A schematic diagram of the possible sites or action of various anti-asthmatic drugs is presented in Fig. 1.
ADRENERGIC STIMULANTS
A very important group of drugs that can bring about symptomatic relief in acute asthmatic attacks are the adrenergic stimulants. There are a variety of these agents, all of which are structural modifications of noepinephrine (1), a neurotransmitter in the adrenergic nervous system. The adrenergic nervous system is one portion of the autonomic nervous system, which regulates or controls the so-called vegetatives or involuntary function of the body.

This nervous system enervates the major organs of the body, eg, the lungs and hearts. The other main segment of the autonomic nervous system is called cholinergic nervous system which has acetylcholine (2) as a neurotransmitter.
The adrenergic and cholinergic nervous systems often have opposing actions upon the tissues in the various organs of the body. The neurotransmitters released by the nerve terminals interact with receptors in the tissue of the various organs of the body to produce specific biological responses. The primary receptors upon which adrenergic stimulants act, have been divided into the -and -adrenergic receptors. The -adr- energic receptors are mainly associated with vasoconstriction, stimulation of uterus, nictitating membrane, pupillary dilation, and intestinal relaxation. The -receptors are associated with smooth muscle relaxation in the lower respiratory tract, relaxation of blood vessels in skeletal muscle, cardiac muscle stimulation, and lipolysis. The receptors associated with responses have been further classified as 1 ( cardiac stimulation and lipolysis) or 2 ( bronchodilation and vasodilation). Recently, there has been considerable effort to develop selective 2 adrenergic stimulants. Such compounds would retain the important bronchodilation activity while being devoid of the cadiovascular, gastrointestinal, and central nervous system side effects produced by compounds possessing significant and 1- adrenergic receptors stimulation.
The adrenergic -stimulants bring about bronchial smooth muscle relaxation by stimulation of adrenergic receptors, which in turn activate adenyl cyclase which catalyzes production of cyclic-3'-5' adenosine monophosphate (cyclic-3',5'-AMP) from adenosine triphosphate (ATP). The cycle-3',5'- AMP then acts as a secondary messenger that eventually produces relaxation of bronchial smooth muscle. Elevated cyclic-3', 5'- AMP is also known to inhibit the release of chemical mediators from the mast cell.
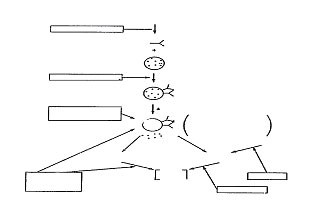
Fig. 1. Schematic diagram of possible sites of action of anti-asthmatic drugs.
A
cetylcholine, along with other cholinergic stimulants, has been shown to increase the level of cyclic-3'5'- guanosine monophosphate (cycle-3', 5'-GMP). Cyclic- 3'5'-AMP is produced from guanosine triphosphate (GTP) by the action of the enzyme guanylate cyclase. Elevated cyclic-3',5' -GMP is thought to induce effects on the lung diametrically opposite to those produced by cycle-3',5',-AMP eg, cycle-3',5'-GMP enhances the release of chemical mediators from the mast cell. Anticholinergic drugs can inhibit the action of acetylcholine and closely related substances in their ability to increase the level of cycle-3', 5'-GMP.
Among the theories of the mechanism of asthmatic attack are those that implicate an abnormality in the autonomic nervous system. Szentivanyi (10) has proposed that asthmatic have an inherited or an acquired deficiency of -adrenergic receptors (adenyl cyclase), thus giving rise to low levels of cyclic-3'-5'-AMP and no bronchodilation when exposed to noxious stimuli. In this proposal neurotransmitters (norepinephrine) and the neurotransmitters (norepinephrine) and neurohormone (epinephrine) (3) that would be released after the challenge of the noxious stimuli, would not be able to bring about the normal homeostatic adjustment due to the lack of, or the improper responsiveness to these -adrenergic receptor stimulants. Thus the bronchial smooth muscle does not respond by relaxation.

Fig. 3. Drugs affecting the level of cyclic-3', 5'-AMP.
Another theory proposes that under stressful circumstances asthmatics have a decreased circulating level of epinephrine, thus again leading to low levels of cyclic-3', 5'-AMP needed for bronchodilation. One adrenergic stimulant used extensively in the therapy of asthma has been isoproternol (7683-59-2). In the past, isoproterenol was popular because of its prompt and intense action in bringing about bronchodilation after inhalation. Unfortunately isoproternol possesses little tissue selectivity and stimulates both 1and 2 adrenergic receptors. The stimulation of the myocardium may lead to undesirable tachycardia, increase in cardiac output and an elevation in blood pressure. Since isoproterenol dilates previously constricted blood vessels in the lung, exaggerating ventilation blood flow inequality may result in a worsening hypoxia, although the airway obstruction is no longer a serious problem. An agent such as phenylephrine (5) may be given in combination with isoproterenol; its -adrenergic activity produces a vasoconstriction to blood vessels; helping to bring about relief from mucosal edema.
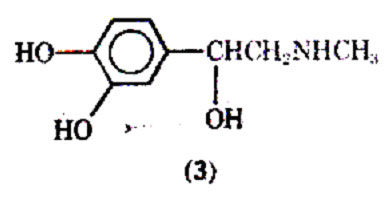
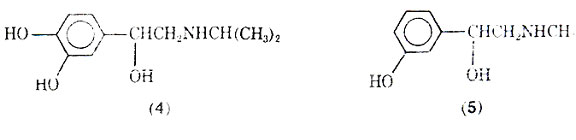
Although isoproterenol has a fast onset of action (2-5 min), and a relatively short duration of action (averaging 1.5-2 h), some patients may become tolerant to its action. The reason for the short duration of action after inhalation is that the drug is well absorbed and rapidly metabolized to 3-O-methylisoproterenol (6) by the enzyme catechol-O methyltransterase (COMT). Since 3-O- methylisoproterenol is a -blocking agent, it has been proposed that this agent once formed could actually inhabit the action of the parent drug molecule isoproterenol; thus, tolerance would develop due to the metabolite. However, doubt has been expressed that 3-O-methylisoproterenol does in fact produce tolerance because of the weak -blocking properties of 3-O- methylisoproterenol. A variety of possible reasons for patients becoming refractory to the effects of isoproterenol and other adrenergic stimulants has been discussed, including the problem of the bronchi becoming plugged with mucus, and the drug's inability to reach the constricted bronchi. Excessive use of pressurized aerosols containing isoproterenol can not only lead to tolerance but may also be associated with an increase in deaths due to excessive 1-stimulation of the heart.
Normal isoproterenol is not effective when taken orally because it is metabolized by the sulphatase enzyme in the intensive to give (7) and by COMT. The sublingual mode of administering isoproterenol has been reported effective in the management of mild asthma attacks.
A well-known agent for treatment of asthma is epinephrine. It was the earliest catecholamine to be used for its bronchodilation activity (8). Epinephrine stimulates both and-adrenergic receptors to about the same degree (25). There are differences in the biological activity of the optical isomers of a number of adrenergic drugs closely related to epinephrine. R-(-)-Epinephrine, the naturally occurring form, is more active than S-(+)- epinephrine on both - and -adrenergic receptors. R-(-)- Epinephrine, which can be extracted from adrenal glands is the isomer used as a drug. Epinephrine can also be synthesized according to the method shown in Fig. 5.
As can be seen, a very similar synthetic process is used for isoproterenol (27-28). Recemic epinephrine is resolved using (+)- tartaric acid to give R-(-)- epinephrine. Epinephrine is generally administered subcutaneosly or intramuscularly and may be given by inhalation. It is not used orally because of its rapid inactivation by metabolism. Once administered by injection or inhalation, the drug produces a rapid bronchodilation with a short duration of action (approximately 1 h). Epinephrine is similar to isoproterenol in that it does not show selectivity in activating -adrenergic receptors. Because of this nonselectivity and its ability to activate - adrenergic receptors, it may produce a variety of side effective including tachycardia, elevated blood pressure, headache, and tremors (8).
Since epinephrine and isoproterenol are catecholamines they are susceptible to oxidation. In the case of epinephrine, eg, oxidation under basic conditions can lead to a reddish-brown material called adrenochrome, which does not possess the biological activity of epiniphrine and should be discarded. An antioxidant such as sodium bisulfite is normally used to stabilize solution of catecholamines such as epinephrine and isoproternol.
Ephedrine (8) a natural product found in Ma Huang has been used for over 5000 years in treating respiratory disorders. The ephedrine used at present in almost exclusively prepared synthetically. Since the ephedrine molecule possesses two asymmetric carbons, there are two possible distereoisomers. The two diastereoisomers forms are ephedrine, possessing an erythro configuration, and pseudo- ephedrine having a threo configuration. Naturally occurring (1R,2S)-(-) ephedrine (8) and (1S,2S)-(+)-pesudo-ephedrine (9) are used clinically for their bronchial relaxant properties.
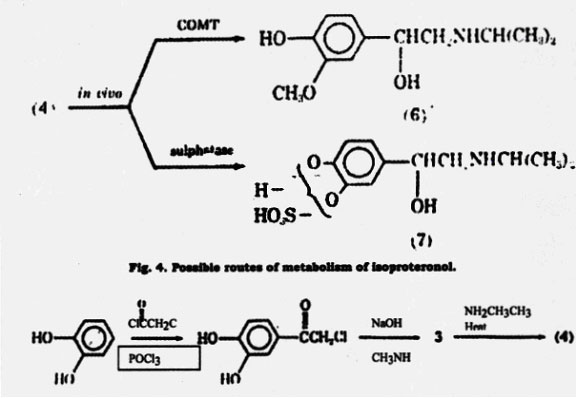
Fig. 5. Synthesis of epinephrine similar to that used for isoproterenol.
Ephedrine is a popular agent used in a host of products for the treatment of bronchial ashma (24,30). Although ephedrine is much less potent than epinephrine, it has the advantage of oral activity and a longer duration of action (3-4 h). The replacement of a catechol ring of epinephrine with a phenyl ring in ephedrine prevents COMT from metabolizing ephedrine. The additional methyl group - to the amino group in ephedrine also prevents metabolism by monoamine oxidase (MAO). The absence of metabolism by these enzymes enhances the oral activity of ephedrine and permits ephedrine to activate adrenergic receptors directly and indirectly. Ephedrine stimulates directly -and -adrengeric reports and by an indirect mechanism it causes release of catecholamines from adrenergic nerve terminals which in turn can activate adrenergic receptors. Thus, although ephedrine does not produce selective bronchodilation, it appears to be a relatively safe drug with minor central nervous system stimulation, pressor, and cardiac side effects. Another compound closely related in chemical structure and in pharmacological action is phenylpropanolamine (700-65-2) (10). It is used for its nasal decongestant and bronchodilator action in a number of proprietary mixtures.
Therapeutically it would be desirable to have an orally active, direct acting 2-adrenergic agonist available with rapid onset and long duration of action. In recent years the search for such new agents has been directed towards chemical modifications of the catecholamine structure. Three general types of modifications have been carried out: (1) replacing or altering the catechol ring system; (2) varying the type of substituent attached to the nitrogen; and (3) altering the phenethylamine side chain.
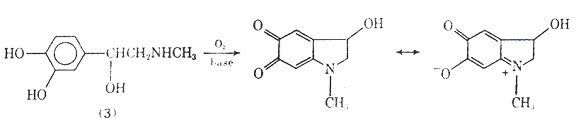
Fig. 6. Oxidation of epinephrine leading to adrenochrome.
Metaproterenol (11) was found to be useful in the treatment of asthma is 1961.
It is analogue of isoproterenol in which the catechol ring system has been replaced by a resorcinol ring system. This change results in a compound that is no longer susceptible to metabolism by COMT and that has a longer duration of action (4h) than isoproterenol. Some selectivity towards 2-adrenergic receptors is observed with metaproterenol, but the compound is 10-40 times less potent than isproternol. This lower potency is, however, compensated by the longer duration of action. Metaproterenol is used both orally and by aerosol inhalation. Apparently some of the selectivity for 2 receptors is lost when the drug is taken orally (8).
Altering the N-substituent of metaproterenol from an isopropyl group to a tert-butyl group gives rise to terbutaline (12) which is a potent, orally active agent with a duration of action up to 7h.
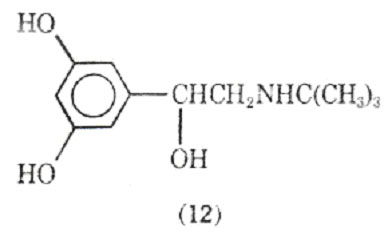
Terbutaline shows selectivity for stimulation of 2-adrenergic receptors over 1-adrenergic tissues. Although less known in North America, it has been extensively investigated in Europe for the treatment of bronchospasmic disorders. In these studies it has been found safe with no evidence of cardiac, hepatic, ranal, or hamatologic changes. Its side effects include increased blood glucose levels and a fine tremor of the fingers. There are - receptors in skeletal muscles and apparently activation of these receptors may be responsible for the tremor.
Increasing the size of the N- substituent of metaproterenol to a 2-(4- hydroxyphenyl) substituent produced a third member of the resorcinol family, fenoteral. This compound is 4 times as potent as metaproterenol as a bronchodilator after inhalation, and shows 2 agonist activity.
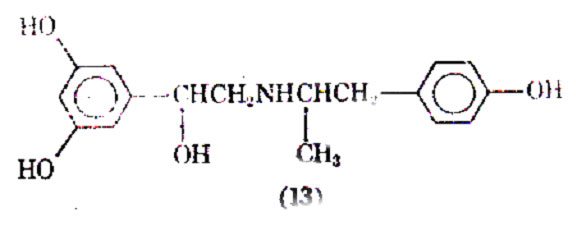
It has been reported as the bronchodilator of choice in the treatment of asthma (8). Other agents belonging to the resorcinol family have been examined for their bronchodilation activity.
Replacing the 3-hdroxyl group with the metabolically more stable hdroxymethylene function produced saligenin derivatives which, it was proposed, would retain some of the electron donating and hydrogen bonding capabilities of the catechol group. Salburamol (14), is the most studied of this series of COMT- resistant compounds (39). It is one fifth as active as isoproterenol in preventing contraction of electrically stimulated isolated guinea pig tracheal muscle and is only a weak 1- agonist in guinea pig atrial muscle. Intravenous administration to animals indicates that sulbutamol is 20-40 times more active than metaproterenol in preventing bronchial muscle contraction caused by acetylcholine or by a suitable vagal stimulation, histamine, 5-hydroxytryptamine, or bradykinin. It is 2-3 times longer acting than isoproterenol and displays no -activity.
Clinically salbutamol has been shown to be a potent and selective bronchodilator when administered orally, intravenously, or by inhalation. At doses required for bronchodilator activity little or no cardiac activity is observed. Its activity has been shown to be prolonged upon aerosol administration, an effect attributed to a decreased absorption of the drug. Metabolically, animal studies indicate that salbutamol is excreted unchanged or as the phenolic glucuronide. Skeletal muscle tremors have been observed as a side effect.
Salmefamol (15) another saligenin derivative has been clinically investigated. It also appears to be a selective 2- agonist with slightly longer duration of action than salbutamol.
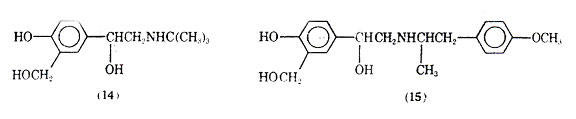
The bronchodilation including both the degree and duration compare favorably with feneterol.
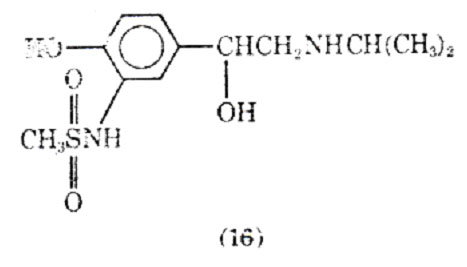
Replacing the 3'-hydraxyl substituent isoproteranol with a methanesulfonamide group led to soterenol (16). It is about epuipotent with isoproterenol at most -receptors, but unlike isoproterenol it also possesses a significant -stimulatory action. In anesthetized cats and dogs soterenol appears to be more active on bronchial muscle than heart muscle (43).
In a recent study in which the 3'-hydroxyl substituent of various catecholamines was substituted with sulfonyl and sulfonylalkyl group, the 3-methylsulfonylmethylene-substituted compound, sulfonterol (17) appears to be a promising new drug. It is about one-half as potent as isoproterenol in guinea pig tracheal relaxation, and one-fifth as potent in decreasing pulmonary resistance in cats; the increase in heart rate that it produces is negligible. The most interesting results appear to be insulfonterol's ability to stimulate selectively the 2- adrenoceptor mediating bronchodilation while mediating vasodepression to a lesser extent. Furthermore, no significant skeletal muscle tremors have been observed. This substance was selected from a number of closely related analogues for clinical trails in man.
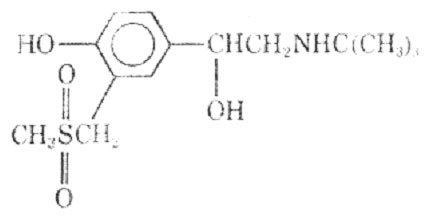
Replacing the 3' -hydroxl groups with a uredio group produced the selective bronchodilator carbuterol (18).
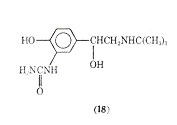
It has been suggested that carbuterol is capable of discriminating between -receptors of the tracheobronchial and vascular smooth muscle. On the basis of its actions in vivo and in vitro the drug was examined for its bronchodilator activity in man.
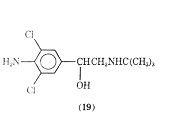
A variety of halogenated phenethylamine derivatives have been investigated for bronchodilation activity. The addition of a chlorine at the 2, 5, or 6 positions of isoproternol gave compounds with no large degree of bronchodiator versus cardiac stimulation specificity although the 2-chloroderivative of isoproterenol is more potent than the parent compound as a brochodilator. As a brochodilator one of the most studied chlorinated phenethylamine derivatives is clenbuterol (19).
It has recently been reported to be a selective - bronchodilator. It appears to have a strong broncholytic response with a long duration of action when administered orally. An oral dose of 10 or 20 µg was found to be similar in bronchodilating potency to an oral dose of 2.5 mg of terbutaline or 20 mg of salbutamol. Clinically clenbuterol has negligible cardiostimulant action and no cardiodepressant activity. It has been reported to have a blocking effect and only slight intrinsic activity on the - receptor.
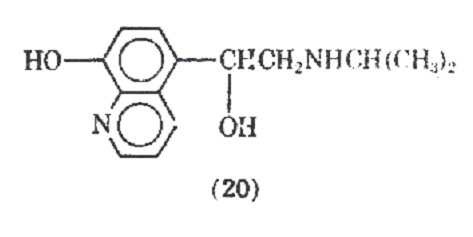
Replacement of the catechol group with an 8-hydroxyquinoline derivative whose chelating properties were to mimic those of the catechol ring led to the preparation of quinterenol (20). It is about 30-50 times less active in vitro on guinea pig trachea and is virtually inactive in the perfused isolated heart of the guinea pig, dog, cat. It has been reported that quinterenol, although selective in its -stimulant actions in vitro, is virtually nonselective in vivo. It is reported to have a slow onset and long duration of action.
Varying the type of substituent attached to the nitrogen, placement of a hexanemoiety between 2 mol of norepinephrine gave hexaprenalice (21).
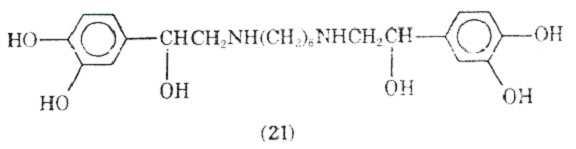
This drug has been used orally and intravenously and appears to be comparable to metaproterenol in its activity. It may have an advantage over other bronchodilators in that it can be used by the intravenous route of administration when significant acidoses is present in the patient. CNS stimulation and mild hypoxemia have been observed as side effects of this bronchodilator.
In a study of tetrahydroisoquinolines, which may be considered cyclized phenethylamine derivatives, Yamato and co-workers found trimetoquinol (22) to be one of the most potent brochodilating agents to date.
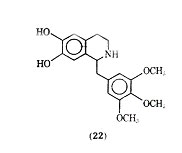
It has been reported that trimetoquinol is a potent stimulant of both 1- and 2 -adrenergic receptors, which is in conflict with earlier work that claimed trimetoquinol to be a selective stimulant of 2 receptors. In a variety of analogues of trimetoquinol it appears that the intact tetrahydroisoquinoline ring system, the 6,7 dihydroxyl group and a large arylmethyl group attached to the 1 position to the tetrahydroisoquinoline ring system are needed for optimum bronchodilation activity.
Trimetoquinol possesses an asymmetric center at the 1 position of the tetrahydroisoquinoline nucleus. When it was resolved, the (-) isomer produced bronchodilation activity twice the recemate's and the (+)-isomer showed little -agonist or antagonist activity. Similar differences have been noted with the optical isomers of other 1-substituted tertrahydroisoquinolines. The (-)-isomer of trimetoquinol has been shown to be an effective bronchodilator when administered by inhalation, parenterally or orally, and to have duration of action longer than with isoproterenol. In a double blind controlled investigation, asthmatic symptoms in children under 3 years of age were reported to be markedly alleviated by trimetoquinol.
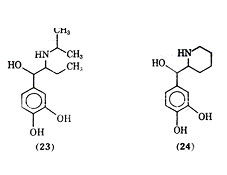
Isoetharine (23) is a -ethyl analogue of isoproterenol. It was shown by lands and co-workers as early as 1950 that isoetharine, although less potent than isoproterenol, produced fewer cardiac side effects for a given degree of bronchodilation. Isoetharine has since been shown to be a selective 2 agonist. Isoetharine possesses two asymmetric centers, and it has been shown that isoetharine is the recemic erythro form of 1-(3,4- dihydroxyphenyl)-2-isopropyl aminobutanol. The (-) erythro isomer of isoetharine has been shown to be the most potent in tracheal relaxation of the four possible isomers. Rimiterol (24), a close analogue of isoetharine with the -side chain substituent and nitrogen substituent forming cyclized side chain moiety, is the erythro form of 3,4- dihydroxyphenyl- (2- piperidyl) - carbinol, a compound with two asymmetric centers. In animal studies rimiterol showed duration of action between those of isoproterenol and salbutamol and appeared to be a selection 2receptor stimulating compound. In clinical studies intravenous injection of rimiterol gave bronchodilation with a duration of action similar to that of isoproterenol and shorter than of salbutamol. Isoproterenol was shown to be about 8 times as potent as rimiterol via inhalation, but rimiterol showed less heart stimulating activity than isoproterenol.
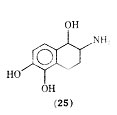
There are recent reports of a tetrahydronaphthalene analogue of isoproterenol in which the side chain has been converted to a fused thing system. The analogue 2-amino-1,2,3,4,- tetrahydro- 1,5,6-naphthalenetriol hydrobromide (25), which is a mixture of cis and trans isomers was found to be equipotent to isoproterenol in vitro in relaxation of guinea pig trachea but considerably less potent on guinea pig atria.
Another approach to producing a long acting selective bronchodilator is the study of prodrugs of catecholamines in animals. In such studies, bitolterol (26) was reported to be a long acting bronchodilator with a significant separation of bronchodilator and cardiovascular actions. Both the oral and aerosol forms of this drug are now being evaluated clinically. The activity of bitolterol is apparently dependent upon its slow hydrolysis to N-tert butylarterenol (27) which then acts as the bronchodilator.
Andrenergic stimulants have an added action besides bronchodilation-the ability to inhibit the release of allergic mediators in lung tissue. This action will be more fully discussed under the section on inhibitors of allergic mediators.

Fig. 7. Bitolterol, a prodrug that gives rise to a bronchodilator.
ANTICHOLINERGICS
One of the agents used for over 100 years in the treatment of asthma is Asthmador. This product contains the naturally occurring stramonium and belladonna alkaloids, one of the principal alkaloids being atropine (28).
The mode of administering Asthmador is through burning the drug and inhaling the fumes. It has been the subject of some drug abuse. The value of atropine and its analogues for relief in respiratory distress has received conflicting reports. The role that anticholinergics play in treating asthma is not well defined at present. Recent work in this area centers around ipratropium bromide (29) which after inhalation produces bronchodilation similar to that obtained with isoproterenol.
It has been suggested that only certain patients may benefit from anticholinergic therapy and that it would be of value to study long-term therapy using anticholinergics such as ipratropium bromide.
INHIBITORS OF THE RELEASE OF ALLERGIC MEDIATORS
A relatively new means of treating bronchial asthma is the use of cromolyn sodium [15826-37-6] (30), a drug developed and introduced in England and in the United Sates.
In 1956, a program was initiated at Fison Corporation to prepare compounds related to khellin (31), a naturally occurring substance having smooth muscle relaxant properties.
The program was directed at preparing drugs possessing the smooth muscle relaxant properties without the nausea and vomiting observed with khellin. In 1965 cromolyn sodium was first prepared. It was found to be inactive as a smooth muscle relaxant but effective in preventing, although not reversing, antigen-induced bronchospasms. Commercially, cromolyn sodium is called Intal, the name derived from interfere and allergy.
The mode of action of cormolyn sodium is unique as it prevents degranulation of mast cells of sensitized tissue in the event of an antigen challenge. This action prevents release of the biochemical mediators of anaphylaxis that bring about the symptoms of asthma, which occur when the body is exposed to an antigen to which it is sensitized. Exposure causes and antigen-antibody reaction. A cascade of events occurs with the eventual release of the biochemical mediators including histamine, slow reacting substances of anaphylaxis (SRS-A), serotonin, and bradykinine.
These mediators interact with receptors in lungs, blood vessels, mucous membranes, etc. Some of the resultant pharmacological actions are bronchial constriction, vasodilation, and increased capillary and mucous membrane permeability. If the immediate hypersensitivity reaction occurs primarily in the bronchial smooth muscle, the result is asthma; if the reaction occurs systemically the result is anaphylactic shock.
In the treatment of asthma, cromolyn sodium does not interfere with the antigen antibody reaction, but appears to inhibit some of the subsequent reactions that eventually lead to the degranulation of pulmonary mast cells. The specific mode of action on the mast cells has been suggested to be through a no specific stabilization of mast cell membranes, through inhibition of enzymes that bring about degranulation, or through prevention of the calcium influx needed to trigger degranulation (83). It is known that cromolyn sodium does not possess any direct antagonism action to the biochemical mediators associated with hypersensitivity reactions, and it has no direct smooth muscle relaxant properties as do the -adrenergic stimulants.
Cromolyn sodium is a prophylactic drug and must be administered before the antigen challenge to the effective Cromolyn sodium has no role in the treatment of an acute asthmatic attack. In clinical tests administering cromolyn sodium after an antigen challenge does not bring about relief in an anaphylactic reaction, and in asthma the drug was considerably less effective just minutes after the antigen challenge. The administration of the drug should be initiated during periods of comparative remission when the lower respiratory system is relatively free from mucus plugging and the patient is able to take the drug through inhalation.
Cromolyn sodium is administered by inhalation with a Spinhaler, the drug being absorbed through the lungs. It is very poorly absorbed from the gastrointestinal tract. Cromolyn sodium (33) is a relatively nontoxic agent generally showing infrequent, mild transient side effects such as nasal decongestion and throat irritation and certain patients may exhibit skin rashes. Because of its low toxicity cromolyn sodium has been used as an adjunct to other drugs. When it is administered with steroids the dose of the steroid can be reduced and fewer side effects from the steroids occur in the treatment of asthma.
The introduction of cromolyn sodium has led toward thinking in terms of prophylaxis rather that crisis therapy of asthma. It serves as a prototype molecule for prophylactic treatment but suffers from the lack of oral activity and limited solubility which recludes aerosol use.
Numerous modification of the structure of cromolyn sodium have been attempted to give an agent with oral activity and longer duration of action. Generally, it has been found-that the bischromone compounds have longer duration of action than the simpler chromone compounds. The synthesis of cromolyn sodium according to Cairns and co-workers is outlined in Fig. 8.
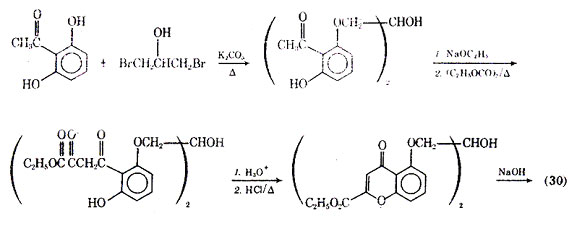
Fig. 8. Synthesis of cromolyn sodium.
Compounds with activity similar to that of cromolyn sodium have been found recently, and their actions have been discussed. It has been proposed that bufrolin [37849-87-9], also called ICI-74, 917 (32), which is chemically unrelated to cromolyn sodium, inhibits allergic reaction by similar mechanisms.
Allergic bronchospasms in guinea pig were inhibited by bufrolin without any evidence of direct bronhodilator activity. Bufrolin was also shown to be 300 times as active as cromolyn sodium against passive cutaneous anaphylaxis (PCA) in the rat. In the PCA test serum, containing antibodies formed in a sensitized animal, is injected intradermally in a normal animal. After a set period of time the perfused area is challenged by intravenous injection of antigen, together with a dye such as Evans Blue, which will leak form the area due to increased capillary permeability resulting from the antigen-antibody reaction. The area of diffusion indicated by the dye is directly related to the intensity of the reaction. Doxantrazole (33), was shown to be effective in inhibiting PCA in a similar manner to cromolyn sodium. The use of dozantrazole orally, in clinical studies with a small number of patients, inhibited allergic asthma attacks to a specific antigen, house dust mite.
Another compound PR-D-92-EA (34) is a potent inhibitor of PCA in rats and prevents immediate hypersensitivity reactions in rats and monkeys. This compound was shown effective in inhibiting the release of the slow reacting substance of anaphylaxis 5,6-Dimethyl-2-nitroindandione (35) was found to be the most active compound in a series of indan-1,3-diones possessing antitiallergenic activity.
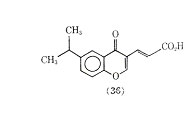
Compound (35) was more than 39 times as active as cromolyn sodium in inhibiting PCA in the rat. In another series of compounds 6-isopropyl-trans-3-(4 oxo-4H-1-benzopyran-3) acrylic acid (36) was also more active than cromolyn sodium in inhibiting PCA. One advantage claimed for (36) is that it has oral activity.
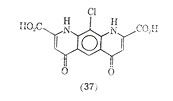
Another compound showing similarly to cromolyn sodium, as inhibitor of mediator release is 10-chloro-1, 4,6,9-tetrahydro-4, 6-dioxopyrido [3,2-g]-quinoline 2,8-dicarboxylic acid (37). It too is reported to have oral activity.
There is a definite advantage with oral drugs in patients with severs airways obstruction where cromolyn sodium inhalation may cease to be effective.
One of the interesting uses of cromolyn sodium has been against ingested allergens. It is reported that cromolyn sodium via inhalation has prevented asthmatic reaction in patients normally sensitive to tomatoes or alcoholic beverages. Fisons has documented that babies who could not tolerated protein from milk were administered cromolyn sodium orally every 6 h for 7 d. After the second day of treatment the babies could tolerate milk.
Adrenergic drugs such as isoproterenol have the ability to block the release of chemical mediators of anaphylaxis. Xanthine derivatives are also reported to have this action. These drugs increase the level of cyclic-3',5'-AMP, and this in turn inhibits the release of chemical mediators of anaphylaxis such as histamine and the slow reacting substance of anaphylaxis. Thus, the adrenergic and xanthine related drugs have in addition to the ability to relax bronchial smooth muscle, the ability to inhibit the release of chemical mediators that bring about the symptoms of asthma.
XANTHINE DERIVATIVES
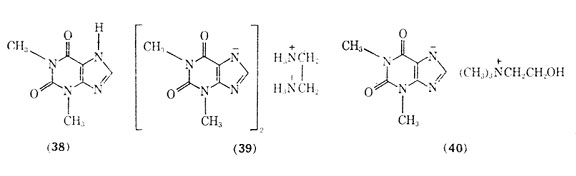
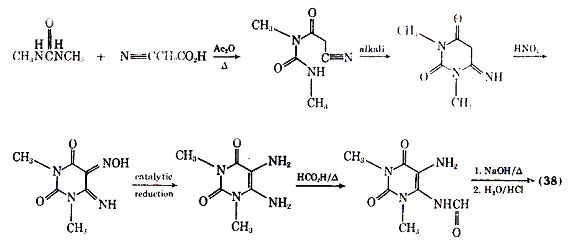
Of the naturally occurring methylated xanthines including theophylline, theobromine, and caffeine, the first compound has been the most effective in treating asthma (see Alkaloids). Theophylline (38), like the adrenergic stimulants, brings about bronchial smooth muscle relaxation. Because of the poor aqueous solubility and gastric irritation of theophylline, a number of salt forms of it have been introduced including aminophylline (theophylline ethylenediamine) (39) and extriphylline (choline salt of theophylline) (40). The most common salt form is aminophylline which was first reported for treatment of status asthmaticus in 1937. The clinical use of aminophylline has been reviewed, and it has been reported to be used extensively as a treatment of severe bronchial asthma.
Theophylline and related xanthines produce bronchial smooth muscle relaxation by increasing cellular levels of cyclic, 3',5'-AMP. However, unlike the adrenergic stimulants which activate adenylcyclase, theophylline inhibits the enzyme phosphodiesterase. The blocked enzyme can no longer hydrolyze cyclic-3',5'-AMP, and levels of this substance increase leading to bronchial relaxation and inhibition of release of chemical mediators of anaphylaxis. Since theophylline acts by a different mechanism in increasing cyclic-3', 5'-AMP than do the adrenergic stimulants, eg, isoproterenol, the administration of aminophylline may produce dramatic results in the management of asthmatic attacks in patients refractory to the adrenergic stimultants. In emergency situations aminophylline is most effective when given by slow intravenous injection. Too rapid an infusion rate may precipitate cardiac arrythmias, hypotension, and convulsions.
Aqueous alcohol solutions of theophylline and its salts have been used to minimize gastric irritation and improve absorption. Orally some of these solutions have provided blood levels of theophylline comparable to intravenous administration within a 30 min period.
Theophylline is found in tea but not in large enough quantities to make it a good source for obtaining the pure drug. It is presently obtained for therapeutic use primarily through total synthesis as outlined in Fig. 9.
A derivative of theophylline that has been used in the treatment of bronchial asthma is dyphylline (dihydroxypropyltheophylline) (41).
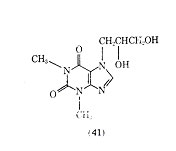
It is 40 times more water-soluble than theophylline and is reported to produce a lower incidence of side effects than theophylline. The half-life of dyphylline (41) after oral administration is 2.1 h, which is shorter than the 3-5 h reported for theophylline. There is some question of the value of dyphylline in producing bronchodilation, however it appears that past
studies have used too low a dose of the drug to see the desired effects. Although a variety of theophylline analogues have been prepared, none of the synthetic compounds show therapeutic advantages over theophylline (105). An interesting fact is that xanthines, eg, theophylline and caffeine, are normally less active in inhibiting the passive cutaneous anaphylactic reaction in the rat than is cromolyn sodium. However, MB 22,948 (42), which was developed by modification of the xanthine nucleus, is 20-50 times as potent as cromolyn sodium in inhibiting a number of test anaphylactic systems, and is orally active.
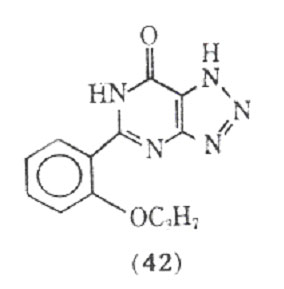
Besides inhibiting the anaphylactic release of mediators from human lung tissue, compound (42) also directly inhibits the effects of histamine and the slow reacting substance of anaphylaxis an isolated human bronchial muscle.
Theophylline has been used in combination with ephedrine in a host of proprietary drugs since 1940. Brown first reported favorable results with the combination therapy, and it has since been suggested for long term therapy of asthma. More recent studies, however, have shown difficulty in measuring the advantages. It now appears that increased toxicity including nervousness, insomnia, and gastrointestinal upset, often results without any additive therapeutic effects.
PROSTAGLANDINS
The prostaglandins are a family of cyclic, oxygenated, C20 fatty acids biosynthesized in human tissue. Derived from polyunsaturated fatty acids by a microsomal synthetase system, they have been associated with a wide range of biological activities at the local level in human tissues and, in particular, with the pathogenesis of the inflammatory response. The prostaglandins are divided into groups (eg, A, B, E, and F) that differ in the type of substitution on the cyclopentane ring and in the number of double bonds in the aliphatic side chains, which is indicated by a numberical subscript after the letter. Of the naturally occurring prostaglandins PGF2 (43), PGE1(44), and PGE2 (45) have been among the most studied in the lower respiratory tract tissues.
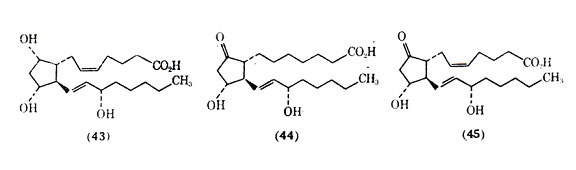
The prostaglandins are quite potent in their ability to alter smooth muscle tone in the lung. Current studies indicate that in isolated human bronchial smooth muscle, relaxation is induced by PGE1 and PGE2 (45) and that contraction is produced by PGF2. The action of protaglandins is of considerable interest in the lung because of speculation that these substances may play an important role in the pathogenesis of asthma. A special characteristic of the prostaglandins is that they are biosynthesized extremely rapidly in a local area rather than being preformed, stored in some state, and then released. The stimuli for local de novo synthesis of protaglandins are under intense investigation. The lifetime of prostaglandins in circulation is very short due to their rapid metabolism. Metabolism of 85-95% of the injected PGE and PGF occurs in the first passage through the vascular bed of the lungs. The evidence for and against prostaglandins playing a role in asthma has been summarized. Asthmatic patients are reported to be more sensitive to the actions of PGF2 than are nonasthmatic patients. One consideration is that a metabolic malfunction can lead to an increase in the level of the bronchoconstrictor PGF2 and a lower level of the bronchodilator PGE2, and that this imbalance in prostaglandins can result in asthma. Another possibility could be a difference in the receptor sensitivities of the prostaglandins in lung tissues resulting in asthma. Although further work needs to be done on the possible role of prostaglandins in asthma, there appears to be little doubt that prostaglandins and chemically related compounds can be of value in treating bronchial asthma.
In clinical studies, PGE1 and PGE2 administered by aerosol are 10 times as potent as isoproterenol in producing bronchodilation in patients with reversible airway obstruction. The prostaglandins are normally shower in onset of action and possess a longer duration of action than isoproterenol. Since PGE1 and PGE2 are natural constituents of the cell, and are metabolized rapidly in the lung, there appear to be few systemic side effects. These prostaglandins also have the ability to increase intracellular cyclic-3',5'-AMP and to prevent the release of chemical mediators of anaphylaxis. Although prostaglandins can increase cyclic-3',5'-AMP, this does not occur through adrenergic receptor stimulation since it has been demonstrated that -adernergic blocking agents do not inhibit the action of prostaglandins. The major side effects of PGE1 and PGE2 administered via the aerosol route are pharyngeal irritation and cough. Given in high does intravenously, PGE2 causes an increase in heart rate and decrease in blood pressure. Since these actions are not observed by inhalation, the aerosol route is the preferred method of administration. However, some instability problems with prostaglandins in solution have been reported.
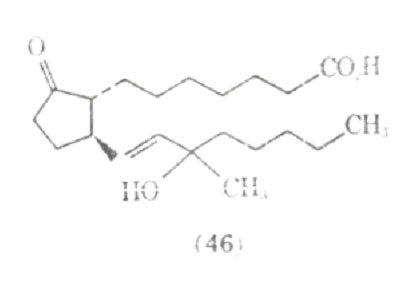
Since the prostaglandins work by a different mechanism than the adrenergic stimulants, it has been suggested that prostaglandins may be of value in treating patients with epinephrine fastness, or situations where the patient is unreactive to the effect of adrenergic stimulants. Work on the structure activity relationship in prostaglandins show that the stereochemistry and the position of functional groups are critical in the pharmacological action produced in the lower respiratory tract. Several synthetic analogues of prostaglandins have been investigated for their bronchodilating activity; one of these compounds is presently undergoing clinical trials as Doxaprost (AY 24,559) (46).
OTHER DRUGS
In addition to the above classes of anti asthmatic, a myriad of drugs has been used in the treatment of asthma. A class of drugs that is often utilized when a patient fails to respond to other agents is the corticosteroids. For example, in status asthmaticus, a severe acute asthmatic attack, if the respiratory symptoms are refractory to other antiasthmatic agents, eg, isoproterenel, then an intravenous corticosteroid may be used to bring about prompt relief. If possible, long term therapy with the corticosteroids would be avoided because of the many possible side effects, including fluid and electrolyte imbalance, peptic ulcer with perforation and hemorrhage, menstrual irregularities, and growth suppression in children (24). At times, however, they are life saving and must be used on a long term basis at the lowest possible dose. The mode of action of the corticosteroids is incompletely understood, but it has been stated that probably the anti-inflammatory action accounts for much of the beneficial effects in asthma (13). Some major advances have been made in administration of therapeutically effective croticosteroids in the treatment of ashma. A variety of topical aerosol steroid preparations are presently being evaluated. The advantage of the aerosolized sprays appears to be the combination of high topical activity in the lung and low systemic activity with relatively short periods of use that avoid many of the untoward reactions of the corticosteroids. Side effects of the aerosolized corticosteroids include local fungal growth, transient hoarseness, and voice changes. Corticotropin [9050-75-3] has shown less marked adrenal suppression and growth retardation in children than the corticosteroids, but it appears to have a limited place in asthma therapy because it must be administered parenterally.
Another class of drugs that might appear to be beneficial in asthma are the antihistamines. However, there are two basic problems:(1) other chemical mediators besides histamine are important in asthma, and the antihistamines are not effective in preventing the action of these other mediators; (2) the drying effect of the antihistamines on mocous membranes in the lung brings about a thicker mucous more difficult to remove. At the present time the antihistamines have no role in the treatment of severe cases of asthma and are generally contraindicated.
Tranquilizers, sedatives, mucolytics, and antibiotics are used in the treatment of asthmatic patients depending upon the psychological and physiological state of the individual.
