The Preparation of Amino Acids and Proteins
10. NATURALLY OCCURRING AMINO ACIDS
As has been pointed out in the earlier part of this chapter, it is necessary for many types of experiments, especially those of a biological nature, that the naturally occurring amino acids be used. In many instances the amino acid is more readily isolated from proteins than by resolution of the synthetic product. For the purpose of obtaining the best yields, it is always desirable to use protein material which is especially rich in the amino acid which is to be isolated. It is not the purpose here to outline in detail the procedures which must be followed in isolating individual amino acids, but rather to present the methods in a general way as well as the references to the literature so that those who may wish to prepare amino acids may do so more readily.
Glycine is best prepared synthetically. Bergmann isolated glycine from gelatin hydrolysates as a complex salt of potassium trioxalatochromiate. According to Towne glycine forms a compound with nitranilic acid [(C2H5O2N)2C6(OH)2 (NO2)2O2)]-which may be used for its quantitative estimation.
The best source for l-cystine is human hair. Wool may also be used. The material is freed from fat by washing with gasoline. It is then hydrolyzed with strong hydrochloric acid until the biuret test is negative. The greater part of the acid is removed by vacuum distillation. Sodium acetate is added until the congo red test is negative. On standing at a low temperature, cystine, some of the tyrosine, and humin separate. The precipitate is filtered, dissolved in weak hydrochloric acid solution, the solution is decolorized by boiling with Norit, and the cystine is precipitated by addition of sodium acetate. The amino acid is washed free from inorganic salts with hot water and dried. The yield from hair is about 5 per cent. Instead of adding sodium acetate in the first step, excess of a suspension of lime may be used. This precipitates the humin while cystine remains in solution. The greater part of the calcium hydroxide in the filtrate is neutralized with hydrochloric acid. Acetic acid is then added to bring the pH of the solution within the isoelectric zone of cystine (pH 3 to 6). The crude cystine is redissolved and reprecipitated as given above. A part of the l-cystine is destroyed during the hydrolysis and some may be racemized. Bennett prefer to hydrolyze the keratin with 50 per cent sulfuric acid. The greater part of the humin is removed by neutralizing the acid with sodium hydroxide to the turning point of congo red and saturating the solution with sodium sulfate. Excess of copper sulfate is added and the solution is again neutralized. The copper precipitate contains cystine, some leucine, aspartic acid, and humic acid. The copper salts are digested with hydrochloric acid and saturated with hydrogen chloride gas. l-Cystine hydrorhloride crystallizes out on standing, while the inactive modifications remain in solution.
d-Glutamic acid is best isolated from the gluten of wheat flour. Since the monosodium salt is produced commercially in large quantities and may be purchased quite cheaply (Ajinomoto, Kin-Saji, Maywesuit), it is more convenient to prepare the amino acid from this source. The free acid is isolated by adding sufficient hydrochloric acid to bring a concentrated solution of the monosodium salt to pH 3.2, the isoelectric point of glutamic acid. The free amino acid crystallizes out at low temperatures. Care must be taken not to convert the glutamic acid to l-pyrrolidone carboxylic acid by heating.
l-Tyrosine may be isolated from either silk waste or casein. If silk is used, hydrolysis is effected by means of hydrochloric acid. As much of the acid as possible is then removed from the filtered solution by vacuum distillation, the solution is decolorized, and the pH is brought to 5.7, the isoelectric point of this amino acid. Crystals arc obtained on permitting the solution to stand in the cold. Purification is effected by recrystallization from water. The yield is 8 to 10 per cent. If casein is used, the protein is dissolved with the aid of ammonia. The mixture is digested with trypsin. A part of the tyrosine will crystallize out without adjusting the pH. The rest is obtained on addition of acetic acid to the isoelectric point of this ammo acid. The remainder of the procedure is the same as that which is given above. The yield of tyrosine is about 4 per cent. When prepared in the manner given above, tyrosine is invariably contaminated with leucine. The latter amino acid may be removed by refluxing the mixture of the two amino acids with glacial acetic acid which dissolves the leucine. l-Diiodotyrosine may be prepared from l-tyrosine according to the procedure of Oswald, l-dichlorotyrosine and l-dibromotyrosine according to the procedure of Zeynek.
l-Tryptophane is best prepared from a tryptic digest of casein. The filtrate, after removal of the greater part of the tyrosine and some of the leucine, may be used for this purpose. The tryptophane is precipitated by the addition of a sulfuric acid solution of mercuric sulfate. Small amounts of tyrosine are removed from the precipitate by washing with a mercuric sulfate solution. The precipitate is suspended in water, made alkaline to phenolphthalein with barium hydroxide, and decomposed with hydrogen sulfide. The barium is precipitated with sulfuric acid, the filtrate is concentrated, and the amino acid is extracted with butyl alcohol. The produce is recrystallized from dilute ethyl alcohol. The yield is about 0.6 per cent.
The isolation of l-methionine has been described. The procedure used by Pirie includes extraction of a casein hydrolysate with butyl alcohol, precipitation of methionine by means of an acetic acid solution of mercuric acetate, and, after decomposition of the mercury compound with hydrogen sulfide, the hydrochloric acid in the solution is neutralized with pyridine. On cooling the solution, methionine crystallizes out. The product obtained contained 95 per cent of methionine and the yield was 1.4 per cent. In the second mentioned procedure casein is digested with pancreatin. After removing tyrosine and tryptophane, additional mercuric sulfate and sulfuric acid are added and the reaction is adjusted to the turning point of congo red by addition of sodium hydroxide solution. The precipitate is filtered, suspended in water, and decomposed by adding barium hydroxide in excess. After removal of barium ions, the methionine is precipitated by addition of mercuric chloride. After decomposing the precipitate with hydrogen sulfide, aniline is added to the concentrated solution in order to neutralize the hydrochloric acid. The methionine is recrystallized from its aqueous solution by addition of alcohol. The yield of analytically pure methionine was about 0.1 per cent. None of these procedures affords a ready method for preparing methionine in large quantities. This is due, in part, to the low content of methionine in casein. The purity of methionine preparations should always be ascertained. The product is often contaminated with phenylalanine and leucine.
The isolation of l-proline from protein hydrolysates, according to the method, depends upon the fact that the copper salts of proline, hydroxyproline, valine, and possibly several other amino acids are soluble in both water and methyl alcohol. On the other hand, certain of the amino acids yield copper salts which are soluble in water but insoluble in methyl alcohol, while the copper salts of the rest are insoluble in both water and methyl alcohol. After decomposing the copper salts, which contain proline, with hydrogen sulfide and concentrating, proline is extracted with absolute alcohol. The alcohol is distilled off and proline is precipitated from the aqueous solution as the picrate. Cox recommend the use of aniline for decomposing proline picrate. If used in excess it will dissolve the aniline picrate and thus afford a way of removing this compound from the aqueous solution of the amino acid. Proline may be extracted directly with absolute alcohol from a gelatin hydrolysate and subsequently converted into the picrate. Either wheat gliadin or gelatin may be used as a source for proline. The yield is nearly quantitative. Bergmann uses ammonium rhodanilate to isolate proline. Arginine is removed from a hydrochloric acid hydrolysate of gelatin and ammonium rhodanilate is added as long as a precipitate is formed. The proline rhodanilate is decomposed by addition of pyridine. From 100 gms. of gelatin 97 gms. of l-proline rhodanilate were obtained. Hydroxyproline may be isolated as the reineckate after removal of arginine and proline. The yield of hydroxyproline is about 10 per cent.
Since l-hydroxyproline also yields a copper salt which is soluble in both water and methyl alcohol, its isolation can be undertaken along with that of proline. The two amino acids can be separated from each other by means of absolute alcohol. Hydroxyproline is isolated as the picrate from the alcohol-insoluble residue. The yield of hydroxyproline, when isolated from gelatin, is about 2 per cent. Both proline and hydroxyproline yield no nitrogen when treated with nitrous acid.
A number of procedures for the isolation of the basic amino acids have been described. Hanke use mercuric chloride for the isolation of l-histidine from a hydrolysate of blood corpuscle paste. The yield from 500 cc. of paste is about 25 gms. of histidine dihydrochloride. Leucine and tyrosine may be isolated as by-products. d-Arginine monohydrochloride may be isolated from a gelatin hydrolysate by means of flavianic acid. Arginine flavianate is decomposed by addition of hydrochloric acid, and the excess of acid beyond that which is required for the formation of arginine monohydrochloride is neutralized with aniline. The yield of arginine monohydrochloride is 7 to 8 per cent. The basic amino acids may also be prepared by the quantitative procedure which is described in Chapter IV.
Perhaps one of the best methods of preparing l-histidine, d-arginine, and d-lysine is by the method of electrical transport which has been discussed in the first section of this chapter. l- Leucine and l-tyrosine may be isolated incidentally. Cox, have described the details of the technique. At pH 5.5 the three hexone bases migrate to the cathode compartment of the three-compartment cell when a direct current is passed through the solution. Arginine and lysine may be separated from histidine by subjecting a solution of the three amino acids to electrical transport at pH 7.5. Arginine is precipitated as the flavianate. The filtrate is freed of flavianic acid by the addition of barium hydroxide, and the excess of barium ions is removed with sulfuric acid. The solution is concentrated, alcohol is added, and lysine picrate crystals are obtained on addition of a solution of picric acid in alcohol. Lysine picrate is decomposed by adding hydrochloric acid. The picric acid is extracted with benzene. Histidine is precipitated from its solution by addition of mercuric sulfate and subsequently isolated in the usual manner as the dihydrochloride. By adjusting the pH to the isoelectric points, leucine and tyrosine may be crystallized out in large part from the protein hydrolysate before subjecting it to electrical transport. The separation of the two amino acids is made by extracting the solids with glacial acetic acid. Cox, King, and Berg report a yield of 51 gm. of histidine monohydrochloride, 21 gm. of arginine monohydrochloride, 62 gm. of lysine chloride, 58 gm. of leucine, and 22 gm. of tyrosine from 4 kilos of blood corpuscle paste. It should be mentioned that in preparing leucine, there is always a possibility that the product obtained is contaminated with methionine or one of the leucine isomers.
l-Aspartic acid may be obtained along with glutamic acid when these amino acids are isolated as the alcohol insoluble barium salts. A more convenient way of preparing aspartic acid is by hydrolysis, if asparagine with dilute hydrochloric or nitric acid. The mineral acid is subsequently neutralized by addition of ammonia to pH 2.8, the isoelectric point of the amino acid, and the aspartic acid is permitted to crystallize. The amino acid may be freed of ammonium salts by extracting it with methyl alcohol and recrystallizing. The yield is 80 to 90 per cent.
d-Alamine can be isolated from silk fibroin. After hydrolysis of the protein, the excess of sulfuric acid is neutralized in the usual manner and tyrosine is crystallized out. The remainder of the amino acids is esterified, glycine ester hydrochloride is removed by crystallization, and the ester of alanine is obtained by fractional distillation. This method possesses the disadvantages which are inherent in the process of esterification and the separation of the amino acid esters.
Thyroxine can be isolated from the thyroid gland by hydrolyzing the protein with barium hydroxide. The racemic form of the amino acid is obtained. Harington has described a method for resolving it into its optically active components. Small amounts of l-thyroxine have been obtained by hydrolyzing the thyroid protein with pepsin and trypsin.
The isolation of serine from sericin has been described by Daft. The protein is hydrolyzed with 25 per cent sulfuric acid. Sufficient barium hydroxide is added to bring the reaction to pH 9.0. After the mixture has stood for one hour the reaction is adjusted to neutrality with sulfuric acid, and the filtrate, after removal of barium sulfate, is concentrated. Crystals of tyrosine, alanine, and serine appear successively. Serine is recrystallized by adding alcohol to the hot aqueous solution of the amino acid.
Serine is racemized by the addition of barium hydroxide to pH 9.0, and it is therefore necessary, in order to obtain l-serine, to resolve the synthetic product. Attempts to isolate the optically active amino acid from the protein hydrolysate have not been particularly successful on account of the solubility of l-serine. Racemic serine is much less soluble than the optically active form. Since l-serine is not isolated directly, the method leaves much to be desired.
The preparation of the other amino acids in considerable amounts from natural sources is in a less satisfactory state. When the naturally occurring form of the amino acid is desired, recourse is usually had to resolution of the synthetic product.
11. PROTEINS
Many of the proteins, both of animal and plant origin, when prepared by different workers, have the same properties and thus appear to be pure compounds. The name of a particular protein, however, does not indicate that, from a chemical standpoint, we are dealing with a single entity. In fact, quite the converse may, at times, be true. For example, it has been shown that casein is a mixture of several proteins. The protamins are likewise not homogeneous chemical compounds, nor is gelatin. Numerous other examples could be cited. On the other hand, as far as our present knowledge extends, some of the animal proteins, such as egg albumin, hemoglobin, and also some of the vegetable proteins, consist of a single type of molecule.
Despite the uncertainties which may exist concerning the homomolecularity of many of the proteins, they can still be used for certain types of experiment. Many of the data which relate to the behavior of proteins in general have been obtained by the use of non-homogeneous proteins. Certain of these proteins possess characteristics, such as an excess of acidic or basic groups, ability to form gels, etc., which make them exceedingly useful for particular experimental purposes.
For physical chemical experiments, it is usually necessary that the protein be salt-free or at least that the content of ash be reduced to a minimum. In the case of proteins which are insoluble at their isoelectric points, it is possible to reduce the ash content to a low level. This may be accomplished, in large part, by washing the isoelectric protein repeatedly with distilled water. Further removal of salts may be effected by electrodialysis. This is carried out by placing the protein in the center compartment of a three-compartment cell, using parchment paper or cellophane to separate the compartments. The cathode and anode compartments contain running distilled water. Electrodes of sheet carbon may be used. On passing a direct current through the cell, the concentration of inorganic electrolytes may be brought to a low level. In the case of proteins which are extracted from natural products by means of a solution of sodium chloride or, in the case of proteins which are precipitated by half or complete saturation with ammonium sulfate, the greater part of the inorganic electrolyte may be removed by placing the protein in a parchment or cellophane bag and dialyzing first against running tap water, arid later against distilled water. Bacterial growth is inhibited by carrying out the dialysis at low temperatures, or by the use of toluol. The dialysis may be expedited by stirring the contents of the bag mechanically. .At times it may be preferable to dialyze the protein solution against a buffer solution whose pH is the same as the isoelectric point of the protein, before dialyzing against distilled water.
It is often desirable to prepare the protein in a dry state. In certain instances this may lead to denaturation. However, if this is not a factor which must be considered, dehydration may be carried out in the following manner. The salt-free precipitate of protein is washed repeatedly, first with 95 per cent alcohol, and finally with, absolute alcohol. The supernatant fluid is removed each time by decantation. Absence of water in the supernatant fluid is determined by testing a portion with anhydrous copper sulfate. The major portion of the alcohol is removed by washing the protein repeatedly with anhydrous ether. The product is quickly filtered on hardened filter paper and dried over sulfuric acid. If care is taken to remove all of the water, the product obtained consists of a light powder resembling flour. If, however, appreciable amounts of water are present, the product will be converted, during the drying procedure, to hard lumps which are difficult to grind. In the case n proteins which crystallize easily, purification may be effected by recrystallization. Insoluble material may be removed by filtration with suction through washed filter paper pulp.
Many of the principles which underline the preparation of the listed below are given in Chapter VII.
Most of the vegetable proteins are obtained from the seeds of plants. The principles which underlie the methods for isolating the vegetable proteins are given. We may illustrate certain of the techniques used in preparing vegetable proteins by using edestin and gliadin as examples. While it is possible to extract edestin from hemp seed without previously removing fat and chlorophyll, it is, however,
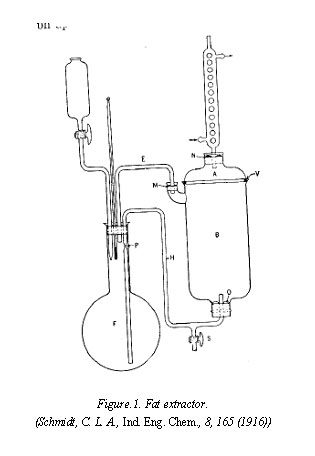
best to remove them. This may conveniently be done by extracting the ground seed with ether, using the extraction apparatus which is shown in Fig.1. The fat-free meal is macerated with 10 volumes of 5 per cent sodium chloride solution at about 50° for an hour and filtered at this temperature through soft filter paper. The reaction of the extract is adjusted to pH 6.9, the isoelectric point of edestin. The solution may dialyzed or permitted to stand in the refrigerator overnight. Crystals of edestin separate. The supernatant fluid is syphoned off, the precipitate is redissolved in warm sodium chloride solution, filtered, and recrystallized. This process is repeated several times. The crystals are washed repeatedly with carbon dioxide-free distilled water at a low temperature. Electrodialysis may be used to reduce the content of ash. The product is finally dehydrated by washing with alcohol and ether and dried over sulfuric acid. The electrically heated vacuum desiccator described may be used for this purpose. According to Osborne the composition of edestin is: C=51.3, H = 6.8, N=18.8, S = 0.9, and O =22.2.
The preparation of gliadin has been described. Wheat flour is moistened with water so as to form a dough. This is washed free from starch by kneading it in running water. The moist gluten is extracted repeatedly with 70 per cent (by volume) alcohol. The filtered extract is concentrated in vacuo below 50°. The concentrated syrup is poured into 5 volumes of one per cent aqueous sodium chloride solution. On shaking, the gliadin is precipitated as a foam. This is dissolved in 70 per cent alcohol. On standing in the refrigerator much of the gliadin separates. The precipitate carries down most of the lipids and suspensoid impurities. It is dissolved and reprecipitated as before. The crude gliadin is finally purified by pouring the concentrated alcohol containing gliadin syrup into a one per cent aqueous solution of lithium chloride and washing the precipitate repeatedly with this solution. The wet gliadin is dissolved in the minimum quantity of alcohol. It is reprecipitated by pouring the solution into four volumes of absolute alcohol, containing 0.025 per cent lithium chloride. The process of dissolving the gliadin in alcohol and precipitating it from a stronger concentration of alcohol is repeated several times. The final precipitate is freed of water by grinding and washing it repeatedly with absolute alcohol and then with ether. It is dried in vacuo. When prepared in this manner, gliadin forms water-clear solutions when dissolved in 50, 60, and 70 per cent alcohol. The nitrogen content of gliadin is about 17.54 per cent, and the ash is about 0.08 per cent. On acid hydrolysis 26.2 per cent of ammonia nitrogen is obtained.
The essential features of the above method are: (a) precipitation of the gliadin in aqueous solution thus permitting thorough washing of the foamy precipitate, and (b) use of lithium chloride instead of sodium chloride to promote precipitation. Since lithium chloride is soluble in both alcohol and ether, it is possible to obtain preparations of gliadin which are practically ash-free. Lithium chloride might well be used to promote precipitation as well as solution of other proteins. By its use one may, at times, dispense with dialysis.
Casein is one of the most useful proteins for certain types of investigative work. One of the more recent descriptions for preparing it is given by Cohn. The casein is precipitated from skimmed milk by addition of 0.05 N HC1, with rapid stirring, to pH 4.6, and the precipitate is washed repeatedly by decanting with distilled water. It is then slowly dissolved by addition of 0.1 N sodium hydroxide to pH 6.3. The solution is filtered and the casein is again precipitated by addition of hydrochloric acid with rapid stirring as before. The precipitate is washed free from chlorides with distilled water and the water is removed by washing with alcohol and ether.
When prepared according to the above procedure, casein consists of at least three, and possibly four, proteins which can be separated from each other by fractionation with suitable solvents. The preparation of other proteins which are present in milk is described by Pedersen.
The preparation of ash-free gelatin is a fairly simple procedure. Granulated gelatin is repeatedly washed with M/128 acetic acid at a low temperature. This is followed by washing with distilled water and alcohol. Another procedure is to wash the granules a number of times, first with a 10 per cent sodium chloride solution containing 5 cc. of concentrated hydrochloric acid per liter, then with weaker concentrations of sodium chloride solution without acid, and finally with distilled water until the wash water is free from electrolytes. This is followed by treatment with alcohol and ether in the usual manner. The use of dilute lithium chloride solution, before washing with distilled water is begun, may facilitate the subsequent removal of the electrolyte. The basis of this procedure is removal of ash from a water-insoluble protein by washing it at the isoelectric point. Since the isoelectric point of gelatin, pH 4.7, lies in the acid region, there is little difficulty in removing such contaminants as calcium phosphate. An analysis of gelatin gave the following percentages: C = 50.5, H = 6.8, N = 17.5, O -25.2.
Gelatin consists of a number of different proteins with molecular weights ranging from 11,000 to 70,000. It has not been possible to separate a definite chemical species from gelatin.
The various methods which have been described for the preparation of egg albumin are essentially minor modifications of the technique which was used by Hopkins. The globulin fraction is precipitated by addition of ammonium sulfate to one half saturation. The egg albumin is crystallized by saturating the solution with ammonium sulfate and adjusting the pH to 4.8, the isoelectric point of this protein, by the addition of dilute sulfuric or acetic acid solution. The electrolyte is removed by dialysis. At least three recrystallizations are necessary in order to remove all of the ovomucoid and conalbunin. The crystals may be washed several times with saturated ammonium sulfate solution containing acetic acid-sodium acetate buffer without appreciable loss of protein. Dialysis may be carried out first against a sodium and potassium phosphate buffer solution, then against distilled water, and finally the last traces of electrolyte may be removed by electro-dialysis. The solution of egg albumin may be kept unaltered by the addition of toluol or by refrigeration. Flosdorf report that this protein, as well as others, may be preserved for long periods of time by their method of rapid freezing and dehydration.
Egg albumin has a nitrogen content of 15.6 per cent. It is one of the proteins which is considered to be homomolecular.
Blood serum yields a number of proteins. Serum globulin may be prepared by adding an equal volume of saturated ammonium sulfate solution to blood serum, centrifuging off the precipitate, dissolving the precipitate in water, and reprecipitating. The last procedure is repeated three or four times. Dialysis is carried out against a phosphate buffer solution of pH 5.5 at 0°. The protein may be kept under toluol in a phosphate buffer solution (0.19 M KH2PO4 and 0.009 M Na2HPO4) at a low temperature. The nitrogen content of horse serum globulin is 15.1 per cent. Serum globulin is a homogeneous protein. It is unstable, easily breaking up into euglobulin and pseudoglobulin. Jameson believe that globulin, euglobulin, and pseudoglobulin represent three phases of a system of the same parent substance, dehydrated globulin. A solution of globulin represents the ordinary isotropic phase, while euglobulin and pseudoglobulin are liquid crystal phases.
Serum albumin is prepared in much the same manner as egg albumin. After removal of the globulin fraction crystallization of the serum albumin may be brought about by the addition of dilute or sulfuric acid dissolved in a solution of ammonium sulfate. The crystals are washed with a dilute solution of ammonium sulfate. They are then dissolved in distilled water and reprecipitated. The recrystallization should be repeated several times. Dialysis is carried out first against a phosphate buffer solution, then against distilled water, and finally electrodialysis is employed to remove the last traces of electrolyte. The nitrogen content of horse serum albumin is 15.6 per cent. Its isoelectric point is about pH 5.5. (106). Serum albumin is a homogeneous protein. Details of the method of preparing serum albumin and serum globulin are given.
In preparing hemoglobin advantage is taken of the fact that certain hemoglobins, viz., those of the horse and dog, are insoluble at the isoelectric point and hence may be easily crystallized. Hemoglobin from the blood of man, the ox, and the sheep is quite soluble in water in its isoelectric condition. It is usually best to wash the red cells repeatedly with isotonic salt solution to free them of serum proteins. The cells are laked by addition of distilled water or distilled water and ether. The stromata are removed by centrifuging. Crystallization may be effected by lowering the temperature to 0° and treating the solution with a mixture of carbon dioxide and oxygen. In methods of this type some of the hemoglobin is altered so that its oxygen-binding capacity is diminished.
In the method of Ross the washed red cells, or a solution of hemoglobin freed from stromata, are subjected directly to electrodialysis. After several hours hemoglobin crystals are formed. Recrystallization with a loss of about 30 per cent may be carried out. The amount of base bound to the hemoglobin may be appreciably reduced by passing a stream of carbon dioxide through the solution during electrodialysis. The hemoglobin is best preserved in the moist state at a low temperature. Even then there is a slow conversion into inactive hemoglobin. Drying leads to a similar change. The concentration of hemoglobin in an aqueous solution may be determined by the method of Stadie. Other methods of preparing hemoglobin are given by Adair. Marshall remove proteins other than hemoglobin from laked blood by the use of aluminum hydroxide. Red blood cells also contain a carbon dioxide-precipitable globulin. Methods for its isolation are given by Bennett.
Methods for preparing globin have been described. The procedure consists in adding hydrochloric acid and acetone containing hydrochloric acid to an aqueous solution of carbon monoxide hemoglobin. A precipitate of globin hydrochloride and solution of heme are obtained. After removal of acetone, the globin powder readily dissolves in water and yields an acidic reaction. Globin may be completely precipitated from this solution by addition of ammonium sulfate to one-third saturation or almost completely precipitated by rapid and complete neutralization. The precipitate has the characteristics of denatured protein. It is insoluble in water at pH 8.0, the isoelectric point of globin. It combines with reduced heme to form hemochromogen.
If the solution of globin hydrochloride is gradually neutralized, the greater part of the globin is converted into a form which is soluble in a neutral solution 0.4 saturated with ammonium sulfate. This form of globin possesses the properties of native globin. It is soluble in distilled water, it can be coagulated by heat, and it can combine with heme to give a product which has the spectrum of lethemoglobin. On reduction, the reduced form of hemoglobin is obtained. When a solution of this product is shaken with air it yields the spectrum of oxyhemoglobin.
Fibrin is prepared by permitting blood to clot and removing the clot, or by whipping the blood and removing the shreds. This material is washed repeatedly in water to remove the soluble proteins. Sodium hydroxide solution is now added and the mixture is warmed to about 60°. After filtration the fibrin is precipitated by addition of acetic or hydrochloric acid until the maximum flocculation occurs. The process of solution and precipitation is repeated several times. The fibrin is finally washed free from electrolytes with distilled water and dehydrated with alcohol and ether. The nitrogen content is 17.2 per cent. The isoelectric region is pH 4.5 to 6.7. Greenberg reports that the electrochemical equivalent of fibrin varies with the mode of preparation.
Fibrinogen may be isolated from either oxalated plasma or from blood to which 25 per cent magnesium sulfate solution (one volume magnesium sulfate solution and four volumes of blood) has been added. The blood is centrifuged and the clear plasma is removed. The fibrinogen is salted out by adding an equal volume of a saturated solution of sodium chloride. The precipitate is washed with distilled water. It is then dissolved in M/6 saline solution and reprecipitated by addition of sufficient solid sodium chloride to make a 15 per cent solution. The electrolyte is finally removed by dialysis. The isoelectric point of fibrinogen is 5.5.
The protamins are present in ripe fish sperm combined with nucleic acid. Clupein is obtained from the sperm of herrings, salmon sperm yields salmin, and sturgeon sperm, sturin. The ripe testicles are shaken with water, the suspension of sperm is strained, and the fluid is coagulated by addition of acetic acid. The precipitate is dehydrated with alcohol and ether. The dried material is extracted repeatedly with a one per cent solution of sulfuric acid and the protamin sulfate is precipitated by addition of alcohol. Further purification is effected by dissolving the protein in water, filtering, and precipitating it by addition of alcohol. The last traces of nucleic acid may be removed by precipitating the protamin with sodium picrate. The picric acid is subsequently removed by addition of sulfuric acid and extraction with ether. The protamin sulfate is precipitated by addition of alcohol.
According to Felix clupein is not a chemical entity but consists of at least four protamins which have been termed A1, A2, B, and C.
It is doubtful that any of the keratins which have been isolated represent homogeneous molecular species. However, for certain types of experimental work, partially purified products are found useful. Goddard prepare wool keratin by dissolving wool in a solution of sodium thioglycolate (pH 12). The solution is filtered and the keratin is precipitated by adding acetic acid. The precipitate is filtered off, ground with acetone which is acidified with hydrochloric acid, washed with ether, and dried. The last traces of thioglycolic acid may be removed by dialysis. The keratin is insoluble in water but can be dissolved by the addition of sodium bicarbonate or sodium carbonate solution. The ability of alkaline sodium sulfide, potassium cyanide, or thioglycolic acid solutions to dissolve keratin appears to depend on the splitting of the disulfide groups which are essential for the maintenance of the fibrous structure of keratin.
Production of protein isolate from groundnut (peanut)
Oilseeds constitute some of the most concentrated forms of food in nature and contain fairly large quantities of protein besides the oil. Among the different constituents of the seed, only the oil has so far been considered of value for human consumption and the oil cake residues left behind after extraction have been used mainly as live-stock feed and manure. Scientific research conducted during the past few decades in this country and elsewhere has shown that the oilseed meal, if properly prepared and processed, can serve as a valuable protein supplement to human dietaries and that the proteins isolated from oilseed meals are likely to find extensive use in specialised food preparations suitable for feeding infants and children.
Isolated vegetable proteins have certain advantages over the parent raw materials in that they are free from: (1) insoluble and indigestible carbohydrates which may swell and interfere in the digestion and utilization of protein particularly in children, (2) odoriferous and bitter principles present in the natural materials which may affect their palatability and (3) trypsin inhibitors, phytates etc., which may affect adversely their nutritive value. Further, protein isolates are 2-4 times as concentrated as the protein source and possess a bland taste, thus permitting ready blending with other natural foodstuffs for increasing their protein content without affecting their palatability.
Processing of screw press groundnut cake for protein isolation
As a source of raw material for the commercial production of groundnut protein, the solvent extracted meal would be the natural choice as this could be processed to yield a fat free protein. Work carried out at the Institute had shown that the screw press cake containing about 9 per cent fat could also be processed to yield a fat-free protein. The method consisted in the removal of fat from the cake dispersion by centrifugal separator thus eliminating the use of solvent extraction in the process. The process was, therefore, studied in greater detail and standardised. The process, is briefly, described below: Raw groundnuts are mildly roasted and decuticled. (Removal of cuticle has been found to be the most effective method of eliminating colour in the groundnut cake and the resulting protein isolate). The decuticled kernels are then pressed in screw-press, yielding a cake containing about 8.5 per cent N and 8-10 per cent fat. The cake is ground to a mesh size of 20-42 (ASTM) and then dispersed in water, keeping the meal to water ratio 1:15. The dispersion is adjusted to the pH range 8.0-8.5; after a peptization period of one hour, it is screened and clarified to obtain the carbohydrate fraction. The clarified protein solution is passed through a cream separator and most of the oil is obtained as a fat-rich emulsion. The protein is precipitated at pH 4.5 from the skimmed solution, the precipitated protein curd washed, centrifuged and dried at 50°C in hot air. The flowÂsheet of the process and yield data are given in Fig.1.
From the flow-sheet, two points emerge: (a) If the fat present in the press meal could he recovered by centrifugation, groundnut kernel itself may be used as raw material and its fat content recovered centrifugally (b) and the yield of protein may be improved by processing the kernel itself.
Integrated processing of groundnut kernel for the separation of oil, protein and carbohydrate meal
When groundnut is dispersed in aqueous medium at optimum pH, the three major constituents namely, oil, protein and starch-fibre tend to separate from one another because of differences in specific gravity. This basic principle has been made use of in the development of the integrated method of processing groundnut which enables the simultaneous separation of oil, protein and the carbohydrate fractions.
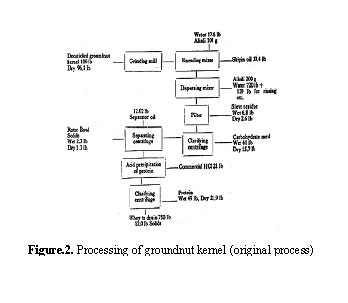
The process, as originally developed, is described as follows: Decuticled (blanched) groundnut kernels are made into a paste and the paste is subjected to 'Skipin' process which consists in adjusting the moisture content of the paste to about 16-20 per cent at 35-37°C. About 30 per cent free oil on the weight of the paste which is liberated at this stage, is drained off and the residual paste made into a dispersion in 8 volumes of water at pH 10.0. The dispersion is clarified to get a carbohydrate meal. The clarified dispersion is passed through a separting centrifuge to obtain another lot of oil equivalent to 12 per cent on the weight of the paste. The remaining skimmed dispersion is acidified to pH 4.5 to obtain the protein curd which is centrifuged and dried. The percentage yields of oil, protein and carbohydrate meal, on dry basis, are 42.0, 21.9 and 15.7 respectively, the oil recovery from the seed amounting to 85 per cent. A flow sheet of the process along with yield data for 100 lb batch size is given in Fig.2.
The process worked with 100 lb batches of the kernel presented the following problems in scaling up:
- The dispersion prepared from the residue after the 'Skipin' process was prone to emulsification rendering its handling difficult. The very first step of clarification itself considerably emulsified the oil making clarification slow and incomplete and the subsequent fat separation very inefficient. The rate of separation of the different constituents was too slow for the process to be feasible on a large scale.
- The 'Skipin' process, necessary as a preliminary step to reduce the fat content of the final emulsion, was a batch process, requiring skilled labour for operation. It also produced some degree of emulsification of the oil.
Consequently, the fat content of the protein isolate could not be reduced to below 10 per cent. Further improvements in the process were, therefore, effected.
Modified Process
The extraction of flaked kernel with alkaline water brings into dispersion most of the protein, about 2/3 of the oil and some starch. The dispersion could be easily handled in continuous nozzle-type centrifuges to separate the protein solu
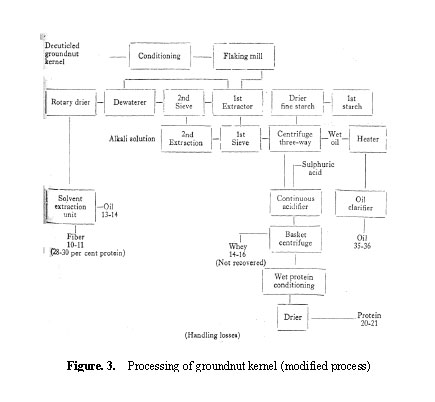
tion, oil and the fine starch. The protein solution is then adjusted to pH 4.5-5.1 and the precipitated protein sedimented and dried. The unextracted residue after dewatering in screw-press is dried and solvent-extracted to recover the remaining oil. The modified process yields a protein of relatively low fat (4 per cent) content and also makes possible the recovery of more oil (91-94 per cent), all of it in a refined state (less than 0.08 FFA). Our recent trials based on an understanding of the factors influencing emulsification of fat globules in the dispersion have indicated the possibility of further reducing the residual fat in the isolate. The flowÂsheet of the modified process and yield data are given in Fig. 3.
The process has the following important features:
- All the operations are capable of being carried out on a continuous basis, reducing thus considerably the time lag between the liberation of protein from the kernel and its being dried as an isolate.
- The oil obtained is in the refined state.
- The rate of separation of the different constituents is high enough to make the process feasible on a large scale.
Comparative analyses of the protein isolates obtained by the different procedures are given in Table I.
TABLE I

Work in progress
Plans are being worked out for setting up a pilot plant with a capacity of 1000 lb of groundnut kernel per hour. This scale of operation would give adequate data to work out the commercial feasibility of the process. Studies are also in progress with regard to the evaluation of the nutritional quality of protein isolate and development of various formulations of protein-rich foods.
The wet processing technique discussed above is being extended to other oil-bearing materials such as cottonseed, sesame, soyabean and coconut. The ultimate object is to make available for human consumption blended compositions based on different vegetable protein isolates.
STUDY OF SOME OF THE OPERATIONS IN THE INTEGRATED PROCESS FOR GROUNDNUTS
An integrated process for groundnuts, developed at this Institute is in some respects analogous to the wet-milling of corn. It aims at the separation of the fat, protein, starch, fiber and water-soluble constituents in a single process. Unlike corn, the groundnut kernel has a high content of fat distributed throughout the kernel which calls for some important variations in the process.
When groundnut is dispersed in water at an alkaline pH, the protein and the water-soluble constituents dissolve, leaving the fat and the insoluble carbohydrates in suspension. By centrifugation the fat is floated away and the carbohydrates sedimented, leaving the protein in the aqueous solution. The protein precipitates out on adjusting the solution to its isoelectric pH, and is sedimented. The water soluble constituents are carried away in the 'whey'. This is the basis of the integrated process. Separation of fat by centrifugation and recovery of protein from the solution, being the two key operations of the process, have been studied in greater detail.
Separation of fat
The separation of fat is dependent on the size of the fat globules in the dispersion. It was therefore considered necessary to study the effect of different operations on the size of fat globules, to avoid those operations that break up the fat globules to small size. The number of fat globules smaller than 2.5 microns diam. was taken as an index of the degree of emulsification caused by the different operations.
The count of fat globules was carried out by centrifuging the sample in a test tube for 5 minutes at 2,250 r.p.m. One ml of the central layer was pipetted out, diluted 50 times with distilled water and adjusted to pH 10. Fat globules, 2.5 microns or less, were counted on five diagonal squares of a haemocytometer, using a microscope (Ã500 magnification). Counts for ten drops were taken for comparison with similar counts on the control. The ratio of the average count of sample to that of the control was expressed as the emulsification ratio.
The following operations were studied: (1) dispersion of the groundnut paste for varying times and at different pH values, (2) comparison of dispersions made from paste subjected to Skipin process with dispersions made directly from the paste, (3) the effect of pumping using centrifugal diaphragm and paddle type pumps, (4) and various types of centrifuges were compared for the degree of fat emulsification produced during the operation of sedimentation of the carbohydrate fraction.
Preliminary experiments revealed that a cage type mixer was preferable to high speed propeller type stirrers. The pH of the dispersion and the time of dispersion had no significant effect on the fat globules. The emulsification produced by the different operations studied is shown in Table I.
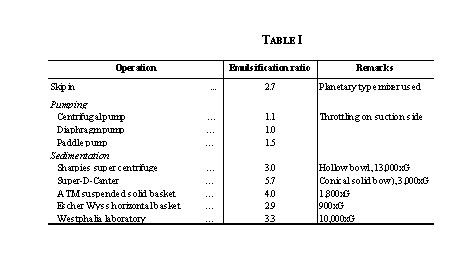
In the Skipin process water is kneaded into the groundnut paste and produced an emulsification ratio of 2.7 The pumps caused no significant emulsification. The process of removing the carbohydrate fraction by sedimentation in a solid-bowl centrifuge produced maximum emulsification in the operations studied. It is, therefore, very desirable to avoid all operations that cause emulsification, before the separation of fat.
Separation of protein
In the recovery of protein, the process of drying affected it more than any other operation. The following aspects of the recovery of protein were studied: (1) precipitation of the protein in a form holding minimum amount of water to reduce drying load, (2) determination of the highest temperature the protein could stand in the wet and dry states, and (3) different methods of drying.
The conditions of protein precipitation are important because they determine the yield and the moisture carried by it. The latter also depends on the efficiency of centrifugation.
The protein was precipitated at pH values between 4.0 and 6.3 and removed by centrifuging in two different types of centrifuges. The moisture content of the sedimented protein and the nitrogen content of the 'whey' were determined to find the optimum conditions of precipitation. The results are shown in Fig.1. The protein was sedimented with the minimum quantity of moisture, when precipitated at pH 5.1, in both types of centrifuges. The loss of protein in the 'whey' was minimum in the range 4.7-6.0. It is therefore concluded that the yield of the protein will be maximum in this range.
The wet protein obtained by centrifuging was subjected to various temperatures (40, 50, 60, 70, 80 and 90°C±1) for 1 hour in a closed dish. Samples were spread in layers and the dish closed to prevent drying, and later freeze-dried. A sample of protein was freeze-dried directly to serve as a control reference. For study of the dry-heat treatment, the wet protein was freeze-dried and the dry samples maintained at temperatures of 80, 90,100,105, 110 and 130°C for three hours. A longer period was used for the dry material as the removal of water in the later stages is slow in the drying process.
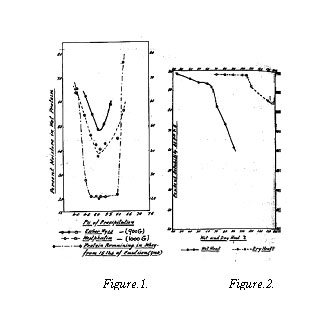
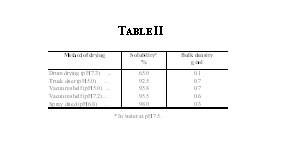
The solubilities in water at pH 7.5, of the variously treated samples of protein and the controls, are shown in Fig. 2. It will be seen that a rapid decrease in solubility is caused at temperatures greater than 60 oC, when the wet protein is subjected to heat. In the dry state, however, even a temperature of 100 oC for three hours does not cause any drastic reduction in solubility. It may, therefore, be concluded from the results that if the wet protein is to be dried in a stream of air, the wet bulb temperature of the air may be raised up to 60°C in the constant rate period, while in the falling rate period the dry bulb temperature may be raised up to about 100°C, without affecting the solubility of the protein to any appreciable extent.
The precipitated protein is capable of being dried in different ways, such as in a truck drier, drum drier, or a spray drier or under vacuum in a shelf drier. Each method has its own advantages and disadvantages. Depending on the end uses of the product one or the other method of drying could be preferred. To select the proper drier, the nature of the product produced by each type of drier must be known. Protein prepared under mild conditions was dried by the different methods and the solubility in water and bulk density measured. The results are given in Table II. The control sample was freeze-dried and had a solubility very close to 100 per cent. The spray drier and the drum drier have the advantages of simple continuous operation that can be easily linked up with any continuous process for the precipitation and sedimentation of protein. Experiments to modify the air-drying process to suit continuous production are under way.
Summary
Emulsification produced by various operations, such as the Skipin process, pumping, centrifugal sedimentation of the carbohydrate fraction etc., on groundnut dispersion in water have been studied. Most of the centrifuges used for the clarification step were found to produce emulsification. Moisture content of protein precipitated at various pH, was determined; pH 5.l is optimum for minimum moisture with maximum yield of protein. The sedimented protein is stable up to 1 hour at 60oC in the wet state or up to l00oC for 3 hours in the dry state. Among the different methods of drying only drum drying reduces the solubility to a considerable extent.
TECHNOLOGY OF PROTEIN-RICH NUTRO FOODS
According to FAO Food Balance Sheets the average daily per capita protein consumption in India is 50.4 g as against the usually recommended level of 60 to 70 g. The potentialities of augmenting the protein intake of the people from untapped edible oil-seed meals are, indeed, great when it is realised that India produces over 7.29 million tons of oil seeds of which groundnut kernels alone account for 2.87 million tons1.
Pioneering investigations carried out at the Central Food Technological Research Institute have led to the establishment of a pilot plant for the production of an acceptable quality of groundnut flour containing 45-50 per cent protein and 5-10 per cent residual oil. Apart from adopting this flour as the major component of Indian Multipurpose Food2'3 and as the chief source of protein in the Tapioca Macaroni products4,5 various other palatable protein-rich foods designated as 'Nutro Foods' have also been developed (Table I). These products comprising Nutro Biscuits, Nutro Macaroni, Nutro Breakfast and Nutro Protein Food for infants and other age groups have all been fortified with calcium and vitamins, the deficiencies of which are widely prevalent in our country. This paper deals with the pilot plant production, shelf-life studies and nutritive value of these
products. Consumer acceptability tests have shown these foods to be palatable and they can, therefore, be produced on a commercial scale.
1. Nutro biscuits: These are sweet biscuits which are very popular in India. But, unlike the customary sweet types, they contain over 16 per cent protein and a

full complement of vitamins-A, D, B1, riboflavin, nicotinic acid and calcium. The high level of protein was achieved by replacing wheat flour in the formulation with groundnut flour to the extent of 40 per cent. Typical batch composition adopted for mechanised production in a modern biscuit factory6 is shown in Table II.
The granularity of groundnut flour was found to influence the properties of dough and texture of the biscuits. If it is as fine as wheat flour (IOXX), water absorption increases, the dough becomes sticky and the biscuits are hard and tend to puff during baking. Less sticky dough and better biscuits were obtained by using slightly coarser flour (about 66 per cent retained on the 100 mesh sieve).
Preparation: Homogeneously blended dry ingredients are mixed with creamed shortening containing the antioxidant, vitamin A, D, glucose syrup, ammonium bicarbonate, sodium bicarbonate and adequate amount of water. Either the rotary embossing machine or the sheeting and punching machines can be used to make biscuits which are baked in a continuous oven at 450-500°F for 5-6 minutes, cooled and packed.
Consumer acceptance: In 1957, the Prime Minister of India donated one lakh of rupees towards the purchase of 1,00,000 lb of Nutro biscuits, which offered a unique opportunity
to organise production on a large scale. About 50 tons of Nutro biscuits were manufactured for distribution by the Indian Red Cross Society, in consultation with the Prime Minister's Relief Fund Organisation, to a number of hospitals, children's schools, welfare centres, earthquake affected areas, and the State Branches of the Indian Red Cross Society. The popularity of the biscuits induced a commercial firm to take up production.
Nutritive value and shelf-life: While developing the formulation, necessary precautions were taken to ensure good shelf-life. This has been confirmed by storage tests7. The biscuits have shown good shelf-life and the over-all nutritive value determined by the growth method has been found satisfactory.
2. Nutro macaroni: Macaroni products in different shapes and sizes are normally made from durum wheat semolina. Use of eggs, milk, gluten fish, and soyabean in certain proportions along with wheat semolina has been reported. Nutro macaroni is based on a blend of wheat semolina (80 per cent) and low fat groundnut flour (20 per cent), with extra fortification with calcium, as calcium carbonate (0.9 per cent), 0.5 mg thiamine hydrochloride, 1.0 mg nicotinic acid, 1.0 mg riboflavin, and 135 I.U. of vitamin D as calciferol.
Pilot plant studies: The product has been made in different shapes, viz., shells, rice, tubes etc., in large quantities in our Macaroni Pilot Plant*. The technique of manufacture is basically similar to that of wheat macaroni. The incorporation of groundnut flour and calcium carbonate was found to affect the consistency of the dough. The water absorption of the dough has to be controlled to ensure proper extrusion.
A study of the effect of processing and the mode of cooking on the nutritive value of the product has shown that the added vitamins are fairly stable. The technique of cooking the product with an appropriate amount of water (1:4) avoids loss of nutrients in the gruel.
Institution feeding trials on 36 subjects, aged 4-12 years, over a period of 24 weeks, were conducted to assess the effect of replacing 50 per cent of rice in their diet by Nutro macaroni. The product was found to be acceptable, and statistically significant gains in weight (Table III) and improvement in nutritional status (Table IV) of children taking Nutro macaroni supplemented diet were recorded.
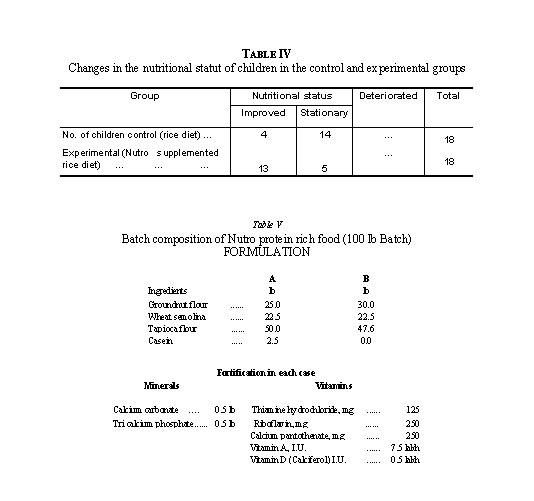
Shelf-life: Systematic storage tests have established that the product has a good shelf-life. No deterioration in the organoleptic quality of the product was observed over a period of 12 months when stored in sealed polythene bags (250 gauge).
3. Nutro protein food: The existing proprietary brands of processed cereal foods for children are beyond the reach of a vast section of Indian families. Products based entirely on milk solids are available at rather high prices. The desirability of having a reasonably priced, nutritionally balanced protein-rich food, adequately fortified with vitamins and minerals, hardly needs any emphasis.
Processing: The technique developed for obtaining suitable dough for macaroni products has been applied successfully for this product also. The ingredients (Table V) are blended thoroughly, prior to doughing with boiling water (30-35 parts). The dough, which is fairly stiff, is vacuum extruded (ATA press), through a die in the shape of 'ringlets' or 'alphabets' which, after passage through a pre-dryer, are steamed for about ten minutes and subsequently dried in a Buhler dryer (Type TTHA). Steaming, though a difficult step in the operation, nevertheless, imparts better flavour and cooking quality to the product.
Nutritive value: The product is being subjected to feeding tests with laboratory animals, and feeding tests on children are also in progress at two centres.
At 10 per cent level of protein intake, the protein efficiency ratio of formulation A and B has been found to be 1.73 and 1.47 respectively. The liver fats averaged 14.9 and 17.5 per cent respectively (on dry basis).
Growth promoting value of the product, when it formed the sole diet having 10 per cent groundnut oil as a supplement, has been determined by rat-growth method. Average weekly gains in weight of 14.3 g and 13.8 g with formulations A and B respectively have been obtained. Experiments to determine the extent to which the use of this product can economise the quantity of milk required for feeding children are in progress.
Acceptability: Laboratory culinary tests suggest that this type of product should find very wide acceptance for the following reasons:
- It takes only 4-5 minutes for cooking.
- The cooked product is more or less bland in taste; it can, therefore, be incorporated into a variety of Indian food preparations.
- Consistent use of this product will ensure supply of much needed proteins, minerals and vitamins.
- It can serve as a very good breakfast food, and when cooked with milk, it becomes very tasty.
4. Nutro breakfast food: This is a porridge-type product having a high protein content (42 per cent) and good flavour and taste. By processing a blend comprising 80 parts groundnut flour, 20 parts wheat flour, and 1-2 per cent malt extract, it has been possible to modify the characteristic taste of groundnut flour.
Process: The dry ingredients are made into a stiff dough, which is cooked under pressure followed by granulation, drying and slight toasting. The contribution of wheat component to flavour development seems specific, because the use of other cereals or starches does not bring about such a modification. Fortification of this product both with vitamins and minerals has to be studied.
vitamin b6 - pyridoxin
1. NOMENCLATURE AND SURVEY
Names:
Pyridoxin: Term suggested by Gyorgy and generally adopted in the United States. Adermin: European name.
Vitamin B6 of Gyorgy.
Anti-acrodynia rat factor.
Rat antidermatitis factor.
Yeast eluate factor.
Factor 1 of Lepkovsky.
Factor Y of Chick.
Vitamin H of Richardson and Hogan and of Booher.
Complimentary factor.
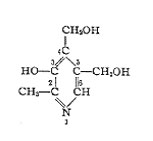
Chemical formula:
Chemical name:
3-Hydroxy-4,5-di(hydroxy-methyI)-2-methyl-pyridine.
2. CHRONOLOGY
1926 GOLDBERGER and LILLIE reported the occurrence of a characteristic dermatitis, called acrodynia, on rats fed a diet deficient in vitamin B2.
1932 OHDAKE in Japan isolated a compound of the formula C8H11O3N.HCl from rice polishings, but failed to recognize its vitamin character.
1934 GYORGY established the difference of the "rat pellagra preventive factor" from vitamin B2 (and vitamin B4) and called the new vitamin "B6."
1938 The isolation of the pure crystalline vitamin B6 was announced independently by five different groups, namely, by LEPKOVSKY, KERESZTESY, and STEVENES, GYORGY, KUHN and WENDT, and ITIBA and MITI.
1939 The chemical structure was elucidated and vitamin B6 was synthesised independently by two groups of workers-by KUHN, WESTPHAL, WENDT and WESTPHAL in Germany and by KERSZTESY, STEVENS, HARRIS, STILLER and FOLKERS in the U.S.A.
3. OCCURRENCE
Vitamin B6 appears to be very widely distributed over the entire animal and plant kingdom. Reliable systematic studies as to the relative quantities in various foodstuffs are scarce. Yeast and rice polishings are especially rich in vitamin B6. Seeds and cereals, for example, wheat and maize, are good sources, especially the germs and the integuments. Molasses, fish and fish livers and mammalian livers contain moderate amounts, and milk, egg yolks, lettuce, spinach, etc., contain small amounts of vitamin B6.
Vitamin B6 occurs in animal and in plant tissues, for example, in yeast and in fish muscle, only to a small extent in the free form. The majority (60-80%) is chemically bound to protein and to starch.
4. ISOLATION
As has been pointed out in the previous section, vitamin B6 occurs to a great extent bound to a protein. From this symplex, vitamin B6 cannot be separated by dialysis. The symplex can, however, be split by heating or by enzymatic hydrolysis.
The free vitamin B6 is extracted with water or with organic solvents such as ether, propanol or butanol. The latter solvents extract less by-products than does water, but continuous extraction is necessary due to the water solubility of the vitamin.
From neutral or acidified water solutions, vitamin B6 can be adsorbed on charcoal and fuller's earth. The absorption on fuller's earth is greatly influenced by the pH of the solution. At pH 5-6 vitamin B6 is not quantitatively absorbed even after three consecutive absorptions. At pH 1 factors Other than B6 are also absorbed. The elution is carried out with barium hydroxide or with butyl alcohol. Vitamin B6 is quantitatively absorbed on zeolite and can be subsequently eluted with 10% potassium chloride. Inert material can be removed with acetone, ethyl alcohol, ethyl acetate, platinum chloride, etc. The vitamin is precipitated by a number of acids, such as by sulfuric acid, phosphotungstic acid, silicotungstic acid, Reinecke's acid, etc. By repeated precipitations, the pure vitamin B6 can be obtained in the form of various salts, such as the hydrochloride. The free base is prepared therefrom by treatment with silver salts.
5. PROPERTIES
Vitamin B6, as a free base, is a colourless crystalline powder, has a slightly bitter taste and melts at 160° C. It is readily soluble in water, in acetone and in alcohol, and slightly soluble in ether and chloroform. Vitamin B6 dialyzes easily. It crystallizes in the form of various salts, for example, as hydrochloride, m. p. 204-206° C. (with decomposition), and as picrate. The hydrochloride is soluble in water (1 g. in 4.5 cc. water) and in alcohol (1 g. in 90 cc. alcohol) and somewhat soluble in acetone. The aqueous solution has a pH of about 3.2. Both the free vitamin B6 and its hydrochloride sublime readily. The hydrochloride is the form in in which this vitamin is marketed. It is a white, odorless powder with salty taste. It is stable to heat, concentrated hydrochloric acid and alkali, but is destroyed by light.
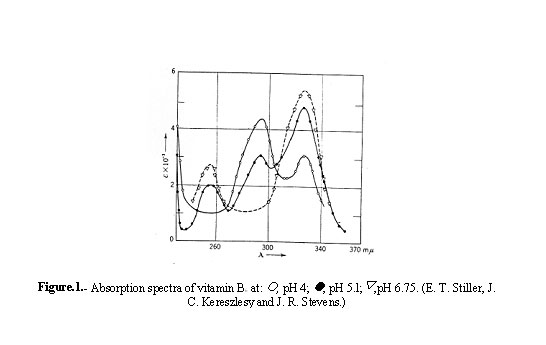
Vitamin B6 is optically inactive. It exhibits a typical ultraviolet absorption spectrum which changes markedly with a change of hydrogen ion concentration. The spectra of the vitamin in aqueous solution between pH 4 and 6.75 are shown in Fig.1.
CONSTITUTION
Vitamin B6 (I) has the empirical formula C8H11O3N and forms salts easily with acids, such as hydrochloric acid, picric acid, etc. The hydrochloride yields an orange-red colour with ferric chloride and couples with diazonium salts, for example, with diazotized sulfanilic acid, which properties suggest the presence of a phenolic or enolic hydroxyl group.
All three oxygens of the molecule are present in the form of hydroxyl groups. A triacetate, which can be distilled, and a tribenzoate (II) can be prepared. An estimation of active hydrogen atoms showed the presence of three in the molecule.
Upon reaction with diazomethane, a compound, C9H13O3N, is obtained, which does not give a colour reaction with ferric chloride and which does not couple with diazotized sulfanilic acid. Treatment of this compound with hydriodic acid yields methyl-iodide. Quantitative estimation of these properties led to the conclusion that a mono-methyl-ether of vitamin B6 (III) is produced by diazomethane. Vitamin B6-methyl-ether is split upon reaction with hydrobromic acid to a hydrobromide of a dibromide, C8H10ONBr3 (IV), which yields vitamin B6 by treatment with silver acetate.
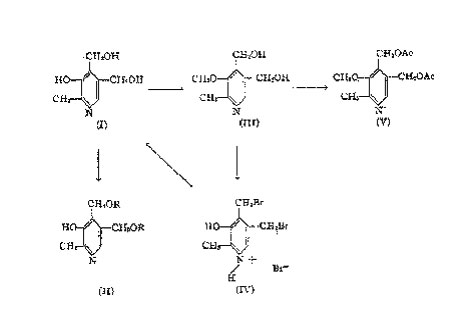
Besides the methyl-ether, diazomethane produces by reaction with vitamin B6 the N-methyl-vitamin B6, which yields a colour reaction with ferric chloride.
The methyl-ether of vitamin B6 is converted by the action of acetic anhydride in pyridine into a diacetyl-methyl-ether (V). Thus it is concluded that of the three hydroxyl groups one is phenolic (or enolic) and the other two are aliphatic hydroxyl groups. All active hydrogen atoms are accounted for and no further active hydrogen atoms could be detected in the diacetyl-methyl-ether. Therefore, it must be concluded that the nitrogen is present in a ring.
The position of the phenolic hydroxyl group was determined, first, by application of the Folin-Denis phenol reagent which produced a colour with vitamin B6 and with b-hydroxy-pyridine, but not with a- and g-hydroxy-pyridine, and second, by investigation of the ultraviolet absorption spectrum which proved to be similar to that of b-hydroxy-pyridine. Upon application of the colour test with 2,6-dichloro-quinone-chlorimide, vitamin B6 gives a blue colour, which effect proves, according to Gibbs, that the p-position to the hydroxyl group is not substituted.
Vitamin B6-methyl-ether does not react with lead tetra-acetate, which indicates that the two aliphatic hydroxyl groups are not in a, b-position. Oxidation in neutral aqueous solution with potassium permanganate in an amount corresponding to two atoms of oxygen converts vitamin B6 methyl-ether (III) into a lactone (VI), which indicates that the two aliphatic hydroxyl groups are either in 1,4- or in 1,5-position to each other. Upon further oxidation of vitamin B6-methyl-ether with barium permanganate, four atoms of oxygen are taken up. The reaction
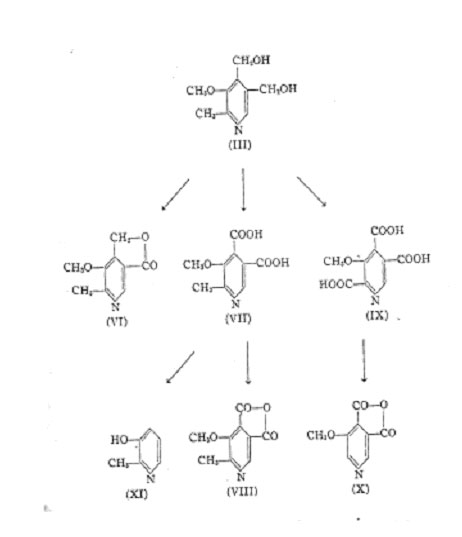
product is a dicarboxylic acid (VII) which contains all the carbon atoms of the vitamin-methyl-ether. By treatment of this dicarboxylic acid with acetic anhydride, the dicarboxylic acid is dehydrated yielding the anhydride (VIII), indicating that the two carboxyl groups are vicinal. Indication of the same fact is given by the fusion of the dicarboxylic acid with resorcinol" which yielded a phthalein having a greenish yellow fluorescence. Decarboxylation of the dibasic acid, by heating the disodium salt with calcium hydroxide, yielded a hydroxy-picoline (XI).
Vitamin B6-methyl-ether on oxidation with potassium permanganate in alkaline solution takes up seven atoms of oxygen and forms a tricarboxylic acid (IX) without the loss of carbon atoms. This tricarboxylic acid yields an anhydride of a dicarboxylic acid (X) while simultaneously losing one mol of carbon dioxide. The tricarboxylic acid yields with ferrous sulfate a reddish colour that is characteristic for pyridine-a-carboxylic acids.
This color reaction is not given by the dicarboxylic acids (VII) and (X). This proves that the carboxyl group lost by anhydration of the tricarboxylic acid was in a-position to the ring-nitrogen and that this carboxyl originated from a methyl group. The existence of the methyl group has also been shown by oxidation of the vitamin chlorohydrate with chromic acid in sulfuric acid whereby acetic acid is obtained.
7. SYNTHESIS
Two different methods have been described for the synthesis of vitamin B6, one of which is a complete synthesis building up the pyridine nucleus from small aliphatic molecules. The other synthesis is a partial degradation of a higher molecular compound to the pyridine derivative of the constitution of vitamin B6.
(a) The Complete Synthesis of Harris and Folkers and of Morii and Makino
By a series of seven reactions vitamin B6 has been synthesized as follows:
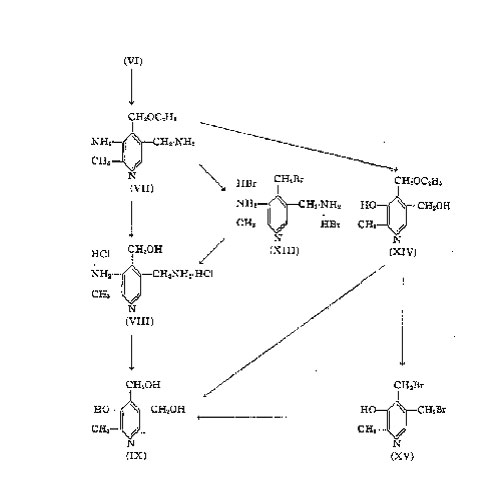
Step 1: Cyano-acetamide (I) is condensed with ethoxy-acetyl-acetone (II) in the presence of piperidine to yield 3-cyano-4-ethoxy-methyl-6-methyl-2-pyridone (III).
Step 2: By nitration of the reaction product of step 1, 3-cyano-4-ethoxy-methyl-5-nitro-6-methyl-2-pyridone (IV) is obtained.
Step 3: Chlorination converts the last-mentioned compound (IV) into 2-methyl 3-3-nitro-4-ethoxy-methyl-5-cyano-6-chloro-pyridine (V).
Step 4: Partial catalytic hydrogenation of (V) yields 2-methyl-3-amino-4-ethoxy-methyI-5-cyano-6-chloro-pyridine (VI).
By a less attractive series of reactions the amino-chloro-compound (VI) can be obtained from the nitro-pyridone (IV) by reduction of the nitro-group to the amino-pyridone (X) followed by chlorination.
Step 5: The cyano-chloro-pyridine (VI) is catalytically hydrogenated to 2-methyl-3-amino-4-ethoxy-methyl-5-amino-methyI-pyridine (VII).
A modification of this step consists in first acetylating the 3-amino-group of (VI) to give (XI) followed by hydrogenation to remove, the chlorine and to convert the cyano-group in the amino-methyl group (XII). Finally the acetyl groups of the 3-amino-group are split off by hydrolysis.
Step 6: The 4-ethoxy-group is now saponified with dilute hydrochloric acid to yield the dihydrochloride of 2-methyl-3-amino-4-hydroxy-methyl-5-amino-methyl-pyridine (VIII).
If this reaction is carried out with hydrobromic acid, the dihydrobromide of a 4-bromo-methyl compound (XIII) is obtained, which must be saponified to give (VIII).
Step 7: The last step of this synthesis consists in diazotization of the di-amine (VIII) to yield vitamin B6 (IX).
Vitamin B6 has also been obtained by a slightly more difficult route, starting with the ethoxy-diamine (VII). By diazotization an ethoxy-dihydroxy-compound (XIV) is obtained, which upon hydrolysis with dilute ydrpchloric acid gives

vitamin B6. When instead of the hydrochloric acid, hydrobromic acid is used, the dibromide (XV) is obtained, which re quires another step to yield finally vitamin B6.
(b) Synthesis by Degradation of Isoquinoline
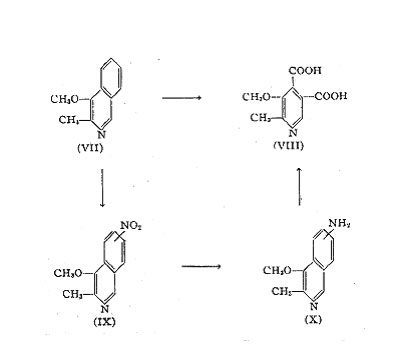
The principle of this synthesis has independently been used by two groups of workers, by Kuhn and co-workers in Germany and by Itiba and Miti in Japan, and consists of the oxidative degradation of 2-methy-3-methoxy-isoquinoline to 2-methyl-3-methoxy-pyridine-4,5-dicarboxyl acid. The isoquinoline derivative is prepared by condensation of potassium phthalimide (I) with a-bromo-propionic acid ester (II) to yield a-phthalimide-propionic acid ester (III), which upon saponification gives 2-methyl-3-hydroxy-isocarbostyril (IV). Chlorination yields the chloro-compound (V) which is converted into the 0-methyl-ether (VI) by methylation with methyl-iodide. The latter compound upon treatment with tin and hydrochloric acid yields the 2-methyl-3-methoxy-isoquinoline (VII).
The 2-methyl-3-methoxy-isoquinoline (VII) is converted into 2-methyl-3-methoxy-pyridine-4,5 dicarboxylic acid (VIII) either by direct oxidation in alkali solution with permanganate or by nitration to a Bz-nitro-compound (IX) followed by reduction to a Bz-amino-compound (X) which is then oxidized with permanganate to yield the dicarboxylic acid (VIII).
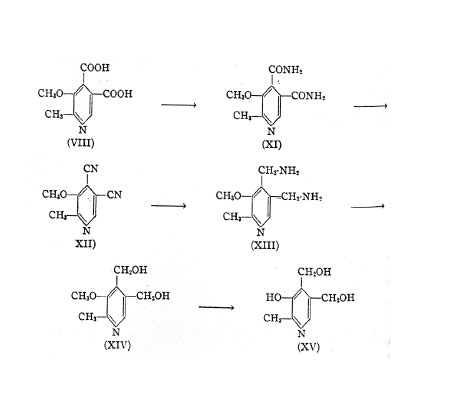
The 2-methyl-3-methoxy~pyridine-4,5-dicarboxylic acid (VIII) is converted through its diamide (XI) into 2-methyl-3-methoxy-4,5-dicyano-pyridine (XII), which by catalytic hydrogenation yields the 2-methyI-3-ethoxy-4,5-diamino-methyl-pyridine (XIII). The latter upon reaction with nitrite gives the vitamin B6
Vitamin B6 O-methyl-ether (XIV). The methyl-ether can be converted into the vitamin B6 (XV) according to one of the previously mentioned methods.
8. INDUSTRIAL METHODS OF PREPARATION
Pure vitamin B6, obtained by extraction of animal or plant material, has never appeared on the market. It has, however, been commercially available in the form of yeast or liver concentrates in mixture with several other members of the vitamin B complex. In the future, extracts of this type will probably be prepared primarily as a source of the less well-known members of the vitamin B-group.
Vitamin B6 is commercially available in the pure crystalline form, synthesized according to both methods outlined in the previous section dealing with the synthesis of vitamin B6.
9. BIOGENESIS
The biogenesis of vitamin B6 is unknown. Theories pertaining to the biogenesis of this vitamin have not been suggested.
10. SPECIFICITY
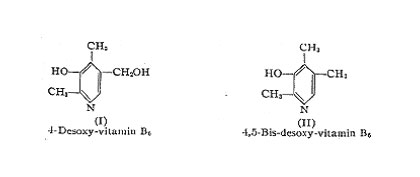
Vitamin B6 apparently owes its physiological action to the molecule as an entity. Compounds of very similar structure and simple derivatives of vitamin B6 are inactive. The di- and triacetate are fully active probably because the organism is able to hydrolyze these esters. The benzoate, however, is inactive. The same is true for the methyl-ether, which, however, shows some activity in a concentration that corresponds to about 500 times the concentration of the free vitamin. 4-Desoxy-vitamin B6 (2,4-dimethyl-3-hydroxy-5-hydroxy-methyI-pyridine (I)) appears to be active in 50 times the concentration of vitamin B6 and 4,5-bis-desoxy-vitamin B6 (2,4,5-trimethyl-3-hydroxy-pyridine (II)) appears to be inactive. A great number of other pyridine derivatives have been tested, but no active compound has been found.
11. DETERMINATION
(a) Chemical Methods
Phenol Test According to Folin and Denis. This test consists in a blue colour given by a phenolic hydroxyl group in the presence of a mixture of sodium tungstate, phosphotungstic acid, phosphoric acid and lithium hydroxide. By the use of this method 0.02-0.08 mg. vitamin B6 can be determined. The colour is measured in a photometer. This method is not specific for vitamin B6.
Cyanine Dye Test. This test can be applied only to the methyl-ether of quaternary vitamin B6 salt. Therefore, vitamin B6 must, first, be converted into the methyl-ether with diazo-methane, and, second, into the iodo-methyl-pyridinium- or a similar pyridinium-compound. These two reactions can be carried out in a yield of 30-40%. The methyl-ether-pyridinium salt, upon heat treatment with chloroform and potassium hydroxide, yields a violet dye of the structure of a carbo-pyridine-cyanine dye (absorption maxima at 599 and 555 mm in alcohol-chloroform). This test is believed to be quite specific to the vitamin since no other a-picoline derivatives are known to occur in plants or animals. By the use of this method 0.8 mg. of pyridoxin can be determined.

Gibb's Phenol Indophenol Test. This test has been adapted to the quantitative colorimetric determination of vitamin B6. A blue colour develops when a vitamin B6 solution is mixed with a veronal buffer (pH 7.6) and a butanol solution of 2,6-dichloroquinone chloroimide. The colour has an absorption maximum at 660 mm and is measured in a colorimeter or spectrophotometer.
Ferric Chloride Method. This method has proved to be of value in the estimation of vitamin B6 in rich sources. The red-brown colour developed by the unknown material is compared with those of standards.
(b) Biological Methods
Yeast Growth Test. Since it has been observed that vitamin B6 stimulates the growth of yeast, a method of determining this vitamin by the rate of growth of yeast has been proposed.
Bacteria Test. Streptobacterium plantarum (Bacterium Acetyl- Cholini Keil 10 S) responds in growth and production of acid to the amount of vitamin B6 present. The increase in growth is measured with a nephelometer.
Rat Growth Test. In this proposed test, the growth rate increase of rats, following the administration of vitamin B6, is determined. Since the decline of the growth rate is not specific for a vitamin B6 deficiency, this test often gives unsatisfactory results.
Acrodynia Rat Test. Vitamin B6 is assayed comparatively on the basis of the percentage incidence of the typical acrodynia type dermatitis in rats in either prophylactic or curative assay procedure.
12. STANDARD
One rat unit vitamin B6 = 10 g, is defined as the amount necessary per day to cure or prevent the typical symptoms of avitaminosis (Gyorgy).
13. PHYSIOLOGY OF PLANTS AND MICROORGANISMS
Although it can be assumed that vitamin B6 is necessary for plants and microorganisms, only few actual facts are known. It has been demonstrated experimentally that vitamin B6 is a nutrilite for yeast and staphylococcus albus and that it is necessary for lactic acid bacteria and for some hemolytic streptococci. Some other bacteria are able to synthesize vitamin B6, for example, those in the rumen of sheep. Only a few observations concerning the vitamin B6 action in higher plants are available. Vitamin B6acts as a growth stimulant on isolated tomato roots and cosmos. The isolated pea root does not require an external supply of vitamin B6 and it must be presumed that the root is able to build up the vitamin from compounds present in the culture medium. Analytical determination of the vitamin B6 content of wheat and maize plants seems to indicate a greater concentration of this vitamin in the germs and the integuments than in the endosperm.
14. ANIMAL PHYSIOLOGY
Free vitamin B6 is rapidly absorbed in the intestines. Vitamin B6 bound to protein material is not absorbed. Plant material, for example, seeds and vegetables, should be cooked to insure full utilization of the vitamin B6 present (as shown in experiments with rats).
Investigations on the behavior of vitamin B6 in the human organism are scarce. Vitamin B6 has not been detected in blood (a reinvestigation seems advisable) but is present in urine, which indicates that this compound is of physiological importance for man. The vitamin is also secreted in milk. When excess amounts of vitamin B6 are administered, they are largely destroyed in the organism. Storage of this vitamin in the organism has not been observed.
Little is known about the physiological action of vitamin B6. It has been suggested that vitamin B6 is connected with the utilization of unsaturated fatty acids. It has also been observed that in vitamin B6-avitaminotic rats, the livers are significantly heavier and contain a higher percentage of total fatty acids, which increase cannot be correlated to the food intake. Rats maintained on a vitamin B6-deficient diet can, according to some investigators, be protected from the symptoms of vitamin B6 deficiency by supplementing the diet with the essential unsaturated fatty acids. Others were unable to obtain this effect but called attention to the very similar type of dermatitis in both vitamin B6 and essential fatty acid deficiency which, however, can be differentiated since edema occurs only in the case of vitamin B6 deficiency. The crux of the problem of the relation of vitamin B6 to the fat metabolism is that the animal organism (tested on rats) needs vitamin B6 for the synthesis of fat from protein. This vitamin is apparently concerned with the metabolism of the amino-acids, as has also been shown in a determination of the protein and carbohydrate appetite in rats by the self-selection method.
The mechanism of the vitamin B6 action is still unknown. Upon the discovery that vitamin B6 occurs in tissues partly bound to proteins, it was suspected that the principal function of this vitamin is to act as part of some enzyme system, like some of the other members of the vitamin B group. The result of an approach to solve this problem was negative: the methyl-iodo-compound of vitamin B6 is not reduced to a dihydro-compound115 in a Banner similar to the reduction of the methyl-iodo-compound of nicotin-amide.
15. AVITAMINOSIS AND HYPOVITAMINOSIS
Vitamin B6 avitaminosis in rats causes a specific symmetrical dermatitis, which is called "acrodynia" and which affects primarily the peripheral parts of the body, such as the paws, the mouth, the tail, the ears and nose, and which is accompanied by edema and scaliness. Furthermore, rats cease growing. In dogs, rats and pigs fits of an epileptiform nature were observed besides the symptoms of subnormal growth and dermatitis. The animals become abnormally excited and any extra stimulus induces fits. The striated and cardiac muscles degenerate, and pathological changes have been noted in the nervous system, especially in the columns of the spinal cord. Chicks also need vitamin B6, but no characteristic dermatitis can be observed during times of vitamin B6 deficiency. The symptoms in chicks consist of slow growth, depressed appetite and inefficient utilization of food. In some cases spastic convulsions and death were observed. In dogs, vitamin B6 deficiency causes a microcytic, hypochromic anemia and in rats a thymus atrophy has been observed. In rats kept on a vitamin B6-deficient diet the accessory organs of reproduction are reduced and the animals show defective sexual behavior.
The present-day knowledge of the action of vitamin B6 in human beings is very limited. The necessity of this vitamin for man has not been proved, but it seems reasonable to assume that vitamin B6is a vitamin in human nutrition. Upon administration of vitamin B6 to pellagrins, recovery has been observed in some cases in which the vitamins B1, B2 and nicotinic acid failed to remove the symptoms. These symptoms include nervousness, insomnia, irritability, cramping pains in the stomach, muscular weakness and muscular rigidity. Patients with pseudohypertrophic muscular dystrophy, for example, respond well to vitamin B6 treatment. Attention has also been called to the possibility that chilblains may afford a clinical demonstration of vitamin B6 deficiency. Promising results have also been obtained in the treatment of myasthenia. Improvement has furthermore been noted in patients with diopathic epilepsy and with macrocytic anemia of pellagra or pernicious anemia in relapse.
(a) Clinical Test Methods
The Urine Test. In urine the excretion of vitamin B6 can be demonstrated for rats and for man. The methods used are either the chemical indophenol test or the biological rat test. The former can be applied to urine when it contains 1 g per cc., but not when less material is present. Urine also contains substances which interfere with the test. In human beings, vitamin B6 can be demonstrated in the urine only after the intake of excessive doses. The normal urinary excretion of man and dogs is less than 0.5 g per cc., while rats excrete about 0.5-1.0 g per cc.
The Blood Test. There is no method known by which vitamin B6 can be determined in blood. As a matter of fact, the presence of this vitamin in blood has not been demonstrated as yet.
16. HYPER VITAMINOSIS
Vitamin B6 is a substance of relatively low toxicity. Chronic toxicity was studied in rats, dogs and monkeys by daily feeding up to 10 mg. per kilogram body weight over periods extending to three months. No significant differences in weight or in the hemoglobin, erythrocytes, leucocytes, etc., were observed. Twenty mg. per kilogram body weight, injected intravenously into cats, had no effect. Single doses up to 1 g. were tolerated without untoward effects. Higher doses produced tonic convulsions and suggest involvement of certain parts of the nervous system. The lethal dose in rats is about 3 g. per kilogram body weight. Vitamin B6 has a sedative effect in man.
17. REQUIREMENTS
Since vitamin B6 has not yet been demonstrated as necessary for human life, no data are available as to the requirements of this vitamin for human beings. In a few clinical cases daily doses of 10-100 mg. were given.
The requirements for rats have been repeatedly investigated and were found to be 10 g daily of the vitamin hydrochloride. Chicks also need this vitamin (about 30 g per day). Sheep, however, do not require an external supply of vitamin B6, since it is synthesized by bacteria in the rumen. The same has also been reported for cattle.
